MIDOSTAURIN
READ …COMPLETE SYNTHESIS AT
Filed under: 0rphan drug status, Phase3 drugs Tagged: MIDOSTAURIN, Orphan Drug, PHASE 3 







MIDOSTAURIN
READ …COMPLETE SYNTHESIS AT
Filed under: 0rphan drug status, Phase3 drugs Tagged: MIDOSTAURIN, Orphan Drug, PHASE 3

Belinostat (PXD101)
PHASE 2, FAST TRACK FDA , ORPHAN STATUS
Belinostat (PXD101) is a novel HDAC inhibitor with IC50 of 27 nM, with activity demonstrated in cisplatin-resistant tumors.
CLINICAL TRIALS…http://clinicaltrials.gov/search/intervention=Belinostat+OR+PXD101
Belinostat inhibits the growth of tumor cells (A2780, HCT116, HT29, WIL, CALU-3, MCF7, PC3 and HS852) with IC50 from 0.2-0.66 μM. PD101 shows low activity in A2780/cp70 and 2780AD cells. Belinostat inhibits bladder cancer cell growth, especially in 5637 cells, which shows accumulation of G0-G1 phase, decrease in S phase, and increase in G2-M phase. Belinostat also shows enhanced tubulin acetylation in ovarian cancer cell lines. A recent study shows that Belinostat activates protein kinase A in a TGF-β signaling-dependent mechanism and decreases survivin mRNA.
| MW 318.07 | |
| MF | C15H14N2O4S |
414864-00-9 cas no
866323-14-0
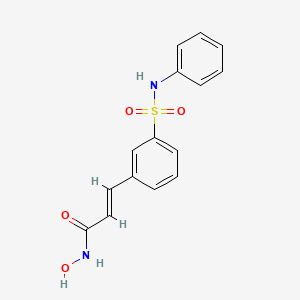
(2E)-N-hydroxy-3-[3-(phenylsulfamoyl)phenyl]acrylamide
A novel HDAC inhibitor
…………………………
Belinostat (PXD101) is experimental drug candidate under development byTopoTarget for the treatment of hematological malignancies and solid tumors. It is a histone deacetylase inhibitor.[1]
A hydroxamate-type inhibitor of histone deacetylase.
NCI: A novel hydroxamic acid-type histone deacetylase (HDAC) inhibitor with antineoplastic activity. Belinostat targets HDAC enzymes, thereby inhibiting tumor cell proliferation, inducing apoptosis, promoting cellular differentiation, and inhibiting angiogenesis. This agent may sensitize drug-resistant tumor cells to other antineoplastic agents, possibly through a mechanism involving the down-regulation of thymidylate synthase
In 2007 preliminary results were released from the Phase II clinical trial of intravenous belinostat in combination with carboplatin and paclitaxel for relapsedovarian cancer.[2] Final results in late 2009 of a phase II trial for T cell lymphomawere encouraging.[3] Belinostat has been granted orphan drug and fast trackdesignation by the FDA.[4]
The study of inhibitors of histone deacetylases indicates that these enzymes play an important role in cell proliferation and differentiation. The inhibitor Trichostatin A (TSA) (Yoshida et al., 1990a) causes cell cycle arrest at both G1 and G2 phases (Yoshida and Beppu, 1988), reverts the transformed phenotype of different cell lines, and induces differentiation of Friend leukaemia cells and others (Yoshida et al., 1990b). TSA (and SAHA) have been reported to inhibit cell growth, induce terminal differentiation, and prevent the formation of tumours in mice (Finnin et al., 1999).
Trichostatin A (TSA)
Suberoylanilide Hydroxamic Acid (SAHA)
Cell cycle arrest by TSA correlates with an increased expression of gelsolin (Hoshikawa et al., 1994), an actin regulatory protein that is down regulated in malignant breast cancer (Mielnicki et al., 1999). Similar effects on cell cycle and differentiation have been observed with a number of deacetylase inhibitors (Kim et al., 1999). Trichostatin A has also been reported to be useful in the treatment of fibrosis, e.g., liver fibrosis and liver cirrhosis. See, e.g., Geerts et al., 1998.
Recently, certain compounds that induce differentiation have been reported to inhibit histone deacetylases. Several experimental antitumour compounds, such as trichostatin A (TSA), trapoxin, suberoylanilide hydroxamic acid (SAHA), and phenylbutyrate have been reported to act, at least in part, by inhibiting histone deacetylase (see, e.g., Yoshida et al., 1990; Richon et al., 1998; Kijima et al., 1993). Additionally, diallyl sulfide and related molecules (see, e.g., Lea et al., 1999), oxamflatin (see, e.g., Kim et al., 1999), MS-27-275, a synthetic benzamide derivative (see, e.g., Saito et al., 1999; Suzuki et al., 1999; note that MS-27-275 was later re-named as MS-275), butyrate derivatives (see, e.g., Lea and Tulsyan, 1995), FR901228 (see, e.g., Nokajima et al., 1998), depudecin (see, e.g., Kwon et al., 1998), and m-carboxycinnamic acid bishydroxamide (see, e.g., Richon et al., 1998) have been reported to inhibit histone deacetylases. In vitro, some of these compounds are reported to inhibit the growth of fibroblast cells by causing cell cycle arrest in the G1 and G2 phases, and can lead to the terminal differentiation and loss of transforming potential of a variety of transformed cell lines (see, e.g., Richon et al, 1996; Kim et al., 1999; Yoshida et al., 1995; Yoshida & Beppu, 1988). In vivo, phenybutyrate is reported to be effective in the treatment of acute promyelocytic leukemia in conjunction with retinoic acid (see, e.g., Warrell et al., 1998). SAHA is reported to be effective in preventing the formation of mammary tumours in rats, and lung tumours in mice (see, e.g., Desai et al., 1999).
The clear involvement of HDACs in the control of cell proliferation and differentiation suggest that aberrant HDAC activity may play a role in cancer. The most direct demonstration that deacetylases contribute to cancer development comes from the analysis of different acute promyelocytic leukaemias (APL). In most APL patients, a translocation of chromosomes 15 and 17 (t(15;17)) results in the expression of a fusion protein containing the N-terminal portion of PML gene product linked to most of RARσ (retinoic acid receptor). In some cases, a different translocation (t(11 ;17)) causes the fusion between the zinc finger protein PLZF and RARα. In the absence of ligand, the wild type RARα represses target genes by tethering HDAC repressor complexes to the promoter DNA. During normal hematopoiesis, retinoic acid (RA) binds RARα and displaces the repressor complex, allowing expression of genes implicated in myeloid differentiation. The RARα fusion proteins occurring in APL patients are no longer responsive to physiological levels of RA and they interfere with the expression of the RA- inducible genes that promote myeloid differentiation. This results in a clonal expansion of promyelocytic cells and development of leukaemia. In vitro experiments have shown that TSA is capable of restoring RA-responsiveness to the fusion RARα proteins and of allowing myeloid differentiation. These results establish a link between HDACs and oncogenesis and suggest that HDACs are potential targets for pharmaceutical intervention in APL patients. (See, for example, Kitamura et al., 2000; David et al., 1998; Lin et al., 1998).
BELINOSTAT
Furthermore, different lines of evidence suggest that HDACs may be important therapeutic targets in other types of cancer. Cell lines derived from many different cancers (prostate, coloreetal, breast, neuronal, hepatic) are induced to differentiate by HDAC inhibitors (Yoshida and Horinouchi, 1999). A number of HDAC inhibitors have been studied in animal models of cancer. They reduce tumour growth and prolong the lifespan of mice bearing different types of transplanted tumours, including melanoma, leukaemia, colon, lung and gastric carcinomas, etc. (Ueda et al., 1994; Kim et al., 1999).
Psoriasis is a common chronic disfiguring skin disease which is characterised by well-demarcated, red, hardened scaly plaques: these may be limited or widespread. The prevalence rate of psoriasis is approximately 2%, i.e., 12.5 million sufferers in the triad countries (US/Europe/Japan). While the disease is rarely fatal, it clearly has serious detrimental effects upon the quality of life of the patient: this is further compounded by the lack of effective therapies. Present treatments are either ineffective, cosmetically unacceptable, or possess undesired side effects. There is therefore a large unmet clinical need for effective and safe drugs for this condition. Psoriasis is a disease of complex etiology. Whilst there is clearly a genetic component, with a number of gene loci being involved, there are also undefined environmental triggers. Whatever the ultimate cause of psoriasis, at the cellular level, it is characterised by local T-cell mediated inflammation, by keratinocyte hyperproliferation, and by localised angiogenesis. These are all processes in which histone deacetylases have been implicated (see, e.g., Saunders et al., 1999; Bernhard et al, 1999; Takahashi et al, 1996; Kim et al , 2001 ). Therefore HDAC inhibitors may be of use in therapy for psoriasis. Candidate drugs may be screened, for example, using proliferation assays with T-cells and/or keratinocytes.
………………………………………………………………………..
PXD101/Belinostat®
(E)-N-hydroxy-3-(3-phenylsulfamoyl-phenyl)-acrylamide, also known as PXD101 and Belinostat®, shown below, is a well known histone deacetylate (HDAC) inhibitor. It is being developed for treatment of a range of disorders mediated by HDAC, including proliferative conditions (such as cancer and psoriasis), malaria, etc.
PXD101 was first described in WO 02/30879 A2. That document describes a multi-step method of synthesis which may conveniently be illustrated by the following scheme.
…………………………………..
GENERAL SYNTHESIS
IGNORE 10

ENTRY 45 IS BELINOSTAT
Scheme 1
By using amines instead of aniline, the corresponding products may be obtained. The use of aniline, 4-methoxyaniline, 4-methylaniline, 4-bromoaniline, 4-chloroaniline, 4-benzylamine, and 4-phenethyamine, among others, is described in the Examples below.
In another method, a suitable amino acid (e.g., ω-amino acid) having a protected carboxylic acid (e.g., as an ester) and an unprotected amino group is reacted with a sulfonyl chloride compound (e.g., RSO2CI) to give the corresponding sulfonamide having a protected carboxylic acid. The protected carboxylic acid is then deprotected using base to give the free carboxylic acid, which is then reacted with, for example, hydroxylamine 2-chlorotrityl resin followed by acid (e.g., trifluoroacetic acid), to give the desired carbamic acid.
One example of this approach is illustrated below, in Scheme 2, wherein the reaction conditions are as follows: (i) RSO2CI, pyridine, DCM, room temperature, 12 hours; (ii) 1 M LiOH or 1 M NaOH, dioxane, room temperature, 3-48 hours; (iii) hydroxylamine 2-chlorotrityl resin, HOAt, HATU, DIPEA, DCM, room temperature, 16 hours; and (iv) TFA/DCM (5:95, v/v), room temperature, 1.5 hours.
Scheme 2
Additional methods for the synthesis of compounds of the present invention are illustrated below and are exemplified in the examples below.
Scheme 3A
Scheme 3B
Scheme 4
Scheme 8
Scheme 9
……………………………………………………………………..
SYNTHESIS
Example 1
3-Formylbenzenesulfonic acid, sodium salt (1)
Oleum (5 ml) was placed in a reaction vessel and benzaldehyde (2.00 g, 18.84 mmol) was slowly added not exceeding the temperature of the reaction mixture more than 30°C. The obtained solution was stirred at 40°C for ten hours and at ambient temperature overnight. The reaction mixture was poured into ice and extracted with ethyl acetate. The aqueous phase was treated with CaC03 until the evolution of C02 ceased (pH~6-7), then the precipitated CaSO4was filtered off and washed with water. The filtrate was treated with Na2CO3 until the pH of the reaction medium increased to pH 8, obtained CaCO3 was filtered off and water solution was evaporated in vacuum. The residue was washed with methanol, the washings were evaporated and the residue was dried in desiccator over P2Oβ affording the title compound (2.00 g, 51%). 1H NMR (D20), δ: 7.56-8.40 (4H, m); 10.04 ppm (1 H, s).
Example 2 3-(3-Sulfophenyl)acrylic acid methyl ester, sodium salt (2)
Sodium salt of 3-formylbenzenesulfonic acid (1) (1.00 g, 4.80 mmol), potassium carbonate (1.32 g, 9.56 mmol), trimethyl phosphonoacetate (1.05 g, 5.77 mmol) and water (2 ml) were stirred at ambient temperature for 30 min., precipitated solid was filtered and washed with methanol. The filtrate was evaporated and the title compound (2) was obtained as a white solid (0.70 g, 55%). 1H NMR (DMSO- dβl HMDSO), δ: 3.68 (3H, s); 6.51 (1 H, d, J=16.0 Hz); 7.30-7.88 (5H, m).
Example 3 3-(3-Chlorosulfonylphenyl)acrylic acid methyl ester (3)
To the sodium salt of 3-(3-sulfophenyl)acrylic acid methyl ester (2) (0.670 g, 2.53 mmol) benzene (2 ml), thionyl chloride (1.508 g, 0.9 ml, 12.67 mmol) and 3 drops of dimethylformamide were added and the resultant suspension was stirred at reflux for one hour. The reaction mixture was evaporated, the residue was dissolved in benzene (3 ml), filtered and the filtrate was evaporated to give the title compound (0.6’40 g, 97%).
Example 4 3-(3-Phenylsulfamoylphenyl)acrylic acid methyl ester (4a)
A solution of 3-(3-chlorosulfonylphenyl)acrylic acid methyl ester (3) (0.640 g, 2.45 mmol) in dichloromethane (2 ml) was added to a mixture of aniline (0.465 g, 4.99 mmol) and pyridine (1 ml), and the resultant solution was stirred at 50°C for one hour. The reaction mixture was evaporated and the residue was partitioned between ethyl acetate and 10% HCI. The organic layer was washed successively with water, saturated NaCl, and dried (Na2S0 ). The solvent was removed and the residue was chromatographed on silica gel with chloroform-ethyl acetate (7:1 , v/v) as eluent. The obtained product was washed with diethyl ether to give the title compound (0.226 g, 29%). 1H NMR (CDCI3, HMDSO), δ: 3.72 (3H, s); 6.34 (1H, d, J=16.0 Hz); 6.68 (1 H, br s); 6.92-7.89 (10H, m).
Example 5 3-(3-Phenylsulfamoylphenyl)acrylic acid (5a)
3-(3-Phenylsulfamoylphenyl)acrylic acid methyl ester (4a) (0.220 g, 0.69 mmol) was dissolved in methanol (3 ml), 1N NaOH (2.08 ml, 2.08 mmol) was added and the resultant solution was stirred at ambient temperature overnight. The reaction mixture was partitioned between ethyl acetate and water. The aqueous layer was acidified with 10% HCI and stirred for 30 min. The precipitated solid was filtered, washed with water and dried in desiccator over P2Os to give the title compound as a white solid (0.173 g, 82%). Example 6 3-(3-Phenylsulfamoylphenyl)acryloyl chloride (6a)
To a suspension of 3-(3-phenylsulfamoylphenyl)acrylic acid (5a) (0.173 g, 0.57 mmol) in dichloromethane (2.3 ml) oxalyl chloride (0.17 ml, 1.95 mmol) and one drop of dimethylformamide were added. The reaction mixture was stirred at 40°C for one hour and concentrated under reduced pressure to give crude title compound (0.185 g).
Example 7
N-Hydroxy-3-(3-phenylsulfamoylphenyl)acrylamide (7a) (PX105684) BELINOSTAT
To a suspension of hydroxylamine hydrochloride (0.200 g, 2.87 mmol) in tetrahydrofuran (3.5 ml) a saturated NaHCOβ solution (2.5 ml) was added and the resultant mixture was stirred at ambient temperature for 10 min. To the reaction mixture a 3-(3-phenylsulfamoylphenyl)acryloyl chloride (6a) (0.185 g) solution in tetrahydrofuran (2.3 ml) was added and stirred at ambient temperature for one hour. The reaction mixture was partitioned between ethyl acetate and 2N HCI. The organic layer was washed successively with water and saturated NaCl, the solvent was removed and the residue was washed with acetonitrile and diethyl ether.
The title compound was obtained as a white solid (0.066 g, 36%), m.p. 172°C. BELINOSTAT
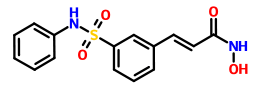
1H NMR (DMSO-d6, HMDSO), δ: 6.49 (1 H, d, J=16.0 Hz); 7.18-8.05 (10H, m); 9.16 (1 H, br s); 10.34 (1 H, s); 10.85 ppm (1 H, br s).
HPLC analysis on Symmetry C18column: impurities 4% (column size 3.9×150 mm; mobile phase acetonitrile – 0.1 M phosphate buffer (pH 2.5), 40:60; sample concentration 1 mg/ml; flow rate 0.8 ml/ min; detector UV 220 nm).
Anal. Calcd for C15Hι4N204S, %: C 56.59, H 4.43, N 8.80. Found, %: C 56.28, H 4.44, N 8.56.
……………………………………………………………………….
SYNTHESIS
US20100286279

…………………………………………………….
SYNTHESIS AND SPECTRAL DATA
Journal of Medicinal Chemistry, 2011 , vol. 54, 13 pg. 4694 – 4720
(E)-N-Hydroxy-3-(3-phenylsulfamoyl-phenyl)-acrylamide (28, belinostat, PXD101).
http://pubs.acs.org/doi/full/10.1021/jm2003552
http://pubs.acs.org/doi/suppl/10.1021/jm2003552/suppl_file/jm2003552_si_001.pdf
The methyl ester (27) (8.0 g) was prepared according to reported synthetic route,
(Watkins, C. J.; Romero-Martin, M.-R.; Moore, K. G.; Ritchie, J.; Finn, P. W.; Kalvinsh, I.;
Loza, E.; Dikvoska, K.; Gailite, V.; Vorona, M.; Piskunova, I.; Starchenkov, I.; Harris, C. J.;
Duffy, J. E. S. Carbamic acid compounds comprising a sulfonamide linkage as HDAC
inhibitors. PCT Int. Appl. WO200230879A2, April 18, 2002.)
but using procedure D (Experimental Section) or method described for 26 to convert the methyl ester to crude
hydroxamic acid which was further purified by chromatography (silica, MeOH/DCM = 1:10) to
afford 28 (PXD101) as off-white or pale yellow powder (2.5 g, 31%).
LC–MS m/z 319.0 ([M +H]+).
1H NMR (DMSO-d6) 12–9 (very broad, 2H), 7.90 (s, 1H), 7.76 (d, J = 7.7 Hz, 1H), 7.70 (d, J
= 7.8 Hz, 1H), 7.56 (t, J = 7.8 Hz, 1H), 7.44 (d, J = 15.8 Hz, 1H), 7.22 (t, J = 7.8 Hz, 2H), 7.08 (d,
J = 7.8 Hz, 2H), 7.01 (t, J = 7.3 Hz, 1H), 6.50 (d, J = 15.8 Hz, 1H);
13C NMR (DMSO-d6) 162.1,
140.6, 138.0, 136.5, 135.9, 131.8, 130.0, 129.2, 127.1, 124.8, 124.1, 121.3, 120.4.
Anal.
(C15H14N2O4S) C, H, N
………………………………………………..
SYNTHESIS
PXDIOI / Belinostat®
(E)-N-hydroxy-3-(3-phenylsulfamoyl-phenyl)-acrylamide, also known as PXD101 and Belinostat®, shown below, is a well known histone deacetylate (HDAC) inhibitor. It is being developed for treatment of a range of disorders mediated by HDAC, including proliferative conditions (such as cancer and psoriasis), malaria, etc.
PXD101 was first described in WO 02/30879 A2. That document describes a multi-step method of synthesis which may conveniently be illustrated by the following scheme.
Scheme 1
Not isolated
ed on (A)
on (D)
d on (H)
There is a need for alternative methods for the synthesis of PXD101 and related compounds for example, methods which are simpler and/or employ fewer steps and/or permit higher yields and/or higher purity product.
Scheme 5
DMAP, toluene
Synthesis 1 3-Bromo-N-phenyl-benzenesulfonamide (3)
To a 30 gallon (-136 L) reactor was charged aniline (2) (4.01 kg; 93.13 g/mol; 43 mol), toluene (25 L), and 4-(dimethylamino)pyridine (DMAP) (12 g), and the mixture was heated to 50-600C. 3-Bromobenzenesulfonyl chloride (1) (5 kg; 255.52 g/mol; 19.6 mol) was charged into the reactor over 30 minutes at 50-600C and progress of the reaction was monitored by HPLC. After 19 hours, toluene (5 L) was added due to losses overnight through the vent line and the reaction was deemed to be complete with no compound (1) being detected by HPLC. The reaction mixture was diluted with toluene (10 L) and then quenched with 2 M aqueous hydrochloric acid (20 L). The organic and aqueous layers were separated, the aqueous layer was discarded, and the organic layer was washed with water (20 L), and then 5% (w/w) sodium bicarbonate solution (20 L), while maintaining the batch temperature at 45-55°C. The batch was then used in the next synthesis.
Synthesis 2 (E)-3-(3-Phenylsulfamoyl-phenyl)-acrylic acid ethyl ester (5)
To the batch containing 3-bromo-N-phenyl-benzenesulfonamide (3) (the treated organic layer obtained in the previous synthesis) was added triethylamine (2.97 kg; 101.19 g/mol; 29.4 mol), tri(o-tolyl)phosphine (119 g; 304.37 g/mol; 0.4 mol), and palladium (II) acetate (44 g; 224.51 g/mol; 0.2 mol), and the resulting mixture was degassed four times with a vacuum/nitrogen purge at 45-55°C. Catalytic palladium (0) was formed in situ. The batch was then heated to 80-900C and ethyl acrylate (4) (2.16 kg; 100.12 g/mol; 21.6 mol) was slowly added over 2.75 hours. The batch was sampled after a further 2 hours and was deemed to be complete with no compound (3) being detected by HPLC. The batch was cooled to 45-55°C and for convenience was left at this temperature overnight.
The batch was then reduced in volume under vacuum to 20-25 L, at a batch temperature of 45-55°C, and ethyl acetate (20 L) was added. The batch was filtered and the residue washed with ethyl acetate (3.5 L). The residue was discarded and the filtrates were sent to a 100 gallon (-454 L) reactor, which had been pre-heated to 600C. The 30 gallon (-136 L) reactor was then cleaned to remove any residual Pd, while the batch in the 100 gallon (-454 L) reactor was washed with 2 M aqueous hydrochloric acid and water at 45-55°C. Once the washes were complete and the 30 gallon (-136 L) reactor was clean, the batch was transferred from the 100 gallon (-454 L) reactor back to the 30 gallon (-136 L) reactor and the solvent was swapped under vacuum from ethyl acetate/toluene to toluene while maintaining a batch temperature of 45-55°C (the volume was reduced to 20-25 L). At this point, the batch had precipitated and heptanes (10 L) were added to re-dissolve it. The batch was then cooled to 0-100C and held at this temperature over the weekend in order to precipitate the product. The batch was filtered and the residue was washed with heptanes (5 L). A sample of the wet-cake was taken for Pd analysis. The Pd content of the crude product (5) was determined to be 12.9 ppm.
The wet-cake was then charged back into the 30 gallon (-136 L) reactor along with ethyl acetate (50 L) and heated to 40-500C in order to obtain a solution. A sparkler filter loaded with 12 impregnated Darco G60® carbon pads was then connected to the reactor and the solution was pumped around in a loop through the sparkler filter. After 1 hour, a sample was taken and evaporated to dryness and analysed for Pd content. The amount of Pd was found to be 1.4 ppm. A second sample was taken after 2 hours and evaporated to dryness and analysed for Pd content. The amount of Pd had been reduced to 0.6 ppm. The batch was blown back into the reactor and held at 40-500C overnight before the solvent was swapped under vacuum from ethyl acetate to toluene while maintaining a batch temperature of 45-55°C (the volume was reduced to 20-25 L). At this point, the batch had precipitated and heptanes (10 L) were added to re-dissolve it and the batch was cooled to 0-100C and held at this temperature overnight in order to precipitate the product. The batch was filtered and the residue was washed with heptanes (5 L). The filtrate was discarded and the residue was dried at 45-55°C under vacuum for 25 hours. A first lot of the title compound (5) was obtained as an off-white solid (4.48 kg, 69% overall yield from 3-bromobenzenesulfonyl chloride (1)) with a Pd content of 0.4 ppm and a purity of 99.22% (AUC) by HPLC.
Synthesis 3 (E)-3-(3-Phenylsulfamoyl-phenyl)-acrvlic acid (6)
To the 30 gallon (-136 L) reactor was charged the (E)-3-(3-phenylsulfamoyl-phenyl)- acrylic acid ethyl ester (5) (4.48 kg; 331.39 g/mol; 13.5 mol) along with 2 M aqueous sodium hydroxide (17.76 L; -35 mol). The mixture was heated to 40-50°C and held at this temperature for 2 hours before sampling, at which point the reaction was deemed to be complete with no compound (5) being detected by HPLC. The batch was adjusted to pH 2.2 using 1 M aqueous hydrochloric acid while maintaining the batch temperature between 40-500C. The product had precipitated and the batch was cooled to 20-300C and held at this temperature for 1 hour before filtering and washing the cake with water (8.9 L). The filtrate was discarded. The batch was allowed to condition on the filter overnight before being charged back into the reactor and slurried in water (44.4 L) at 40-500C for 2 hours. The batch was cooled to 15-20°C, held for 1 hour, and then filtered and the residue washed with water (8.9 L). The filtrate was discarded. The crude title compound (6) was transferred to an oven for drying at 45-55°C under vacuum with a slight nitrogen bleed for 5 days (this was done for convenience) to give a white solid (3.93 kg, 97% yield). The moisture content of the crude material was measured using Karl Fischer (KF) titration and found to be <0.1% (w/w). To the 30 gallon (-136 L) reactor was charged the crude compound (6) along with acetonitrile (47.2 L). The batch was heated to reflux (about 80°C) and held at reflux for 2 hours before cooling to 0-10°C and holding at this temperature overnight in order to precipitate the product. The batch was filtered and the residue was washed with cold acetonitrile (7.9 L). The filtrate was discarded and the residue was dried under vacuum at 45-55°C for 21.5 hours. The title compound (6) was obtained as a fluffy white solid (3.37 kg, 84% yield with respect to compound (5)) with a purity of 99.89% (AUC) by HPLC.
Synthesis 4 (E)-N-Hvdroxy-3-(3-phenylsulfamoyl-phenyl)-acrylamide (PXD101) BELINOSTAT
To the 30 gallon (-136 L) reactor was charged (E)-3-(3-phenylsulfamoyl-phenyl)-acrylic acid (6) (3.37 kg; 303.34 g/mol; 11.1 mol) and a pre-mixed solution of 1 ,8-diazabicyclo[5.4.0]undec-7-ene (DBU) in isopropyl acetate (IPAc) (27 g in 30 L; 152.24 g/mol; 0.18 mol). The slurry was stirred and thionyl chloride (SOCI2) (960 mL; density ~1.631 g/mL; 118.97 g/mol; -13 mol) was added to the reaction mixture and the batch was stirred at 20-300C overnight. After 18.5 hours, the batch was sampled and deemed to be complete with no compound (6) being detected by HPLC. The resulting solution was transferred to a 100 L Schott reactor for temporary storage while the
30 gallon (-136 L) reactor was rinsed with isopropyl acetate (IPAc) and water. Deionized water (28.9 L) was then added to the 30 gallon (-136 L) reactor followed by 50% (w/w) hydroxylamine (6.57 L; -1.078 g/mL; 33.03 g/mol; -214 mol) and another charge of deionized water (1.66 L) to rinse the lines free of hydroxylamine to make a 10% (w/w) hydroxylamine solution. Tetrahydrofuran (THF) (6.64 L) was then charged to the
30 gallon (-136 L) reactor and the mixture was stirred and cooled to 0-100C. The acid chloride solution (from the 100 L Schott reactor) was then slowly charged into the hydroxylamine solution over 1 hour maintaining a batch temperature of 0-10°C during the addition. The batch was then allowed to warm to 20-300C. The aqueous layer was separated and discarded. The organic layer was then reduced in volume under vacuum while maintaining a batch temperature of less than 300C. The intention was to distill out 10-13 L of solvent, but this level was overshot. A larger volume of isopropyl acetate (IPAc) (16.6 L) was added and about 6 L of solvent was distilled out. The batch had precipitated and heptanes (24.9 L) were added and the batch was held at 20-30°C overnight. The batch was filtered and the residue was washed with heptanes (6.64 L). The filtrate was discarded and the residue was dried at 45-55°C under vacuum with a slight nitrogen bleed over the weekend. The title compound (PXD101) was obtained as a light orange solid (3.11 kg, 89% yield with respect to compound (6)) with a purity of 99.25% (AUC) by HPLC.
The title compound (PXD101) (1.2 kg, 3.77 mol) was dissolved in 8 volumes of 1:1 (EtOH/water) at 600C. Sodium bicarbonate (15.8 g, 5 mol%) was added to the solution. Water (HPLC grade) was then added at a rate of 65 mL/min while keeping the internal temperature >57°C. After water (6.6 L) had been added, crystals started to form and the water addition was stopped. The reaction mixture was then cooled at a rate of 10°C/90 min to a temperature of 0-10cC and then stirred at ambient temperature overnight. The crystals were then filtered and collected. The filter cake was washed by slurrying in water (2 x 1.2 L) and then dried in an oven at 45°C for 60 hours with a slight nitrogen bleed. 1.048 kg (87% recovery) of a light orange solid was recovered. Microscopy and XRPD data showed a conglomerate of irregularly shaped birefringant crystalline particles. The compound was found to contain 0.02% water.
As discussed above: the yield of compound (5) with respect to compound (1) was 69%. the yield of compound (6) with respect to compound (5) was 84%. the yield of PXD101 with respect to compound (6) was 89%.
……………….
FORMULATION
Formulation Studies
These studies demonstrate a substantial enhancement of HDACi solubility (on the order of a 500-fold increase for PXD-101) using one or more of: cyclodextrin, arginine, and meglumine. The resulting compositions are stable and can be diluted to the desired target concentration without the risk of precipitation. Furthermore, the compositions have a pH that, while higher than ideal, is acceptable for use.
UV Absorbance
The ultraviolet (UV absorbance E\ value for PXD-101 was determined by plotting a calibration curve of PXD-101 concentration in 50:50 methanol/water at the λmax for the material, 269 nm. Using this method, the E1i value was determined as 715.7.
Methanol/water was selected as the subsequent diluting medium for solubility studies rather than neat methanol (or other organic solvent) to reduce the risk of precipitation of the cyclodextrin.
Solubility in Demineralised Water
The solubility of PXD-101 was determined to be 0.14 mg/mL for demineralised water. Solubility Enhancement with Cvclodextrins
Saturated samples of PXD-101 were prepared in aqueous solutions of two natural cyclodextrins (α-CD and γ-CD) and hydroxypropyl derivatives of the α, β and Y cyclodextrins (HP-α-CD, HP-β-CD and HP-γ-CD). All experiments were completed with cyclodextrin concentrations of 250 mg/mL, except for α-CD, where the solubility of the cyclodextrin was not sufficient to achieve this concentration. The data are summarised in the following table. HP-β-CD offers the best solubility enhancement for PXD-101.
Phase Solubility Determination of HP-β-CD
The phase solubility diagram for HP-β-CD was prepared for concentrations of cyclodextrin between 50 and 500 mg/mL (5-50% w/v). The calculated saturated solubilities of the complexed HDACi were plotted against the concentration of cyclodextrin. See Figure 1.
………………………..

| US2008274120 | 11-7-2008 | Histone Deacetylase (Hdac) Inhibitors (Pxd101) for the Treatment of Cancer Alone or in Combination With Chemotherapeutic Agent |
| US2008227845 | 9-19-2008 | CYCLOOXYGENASE-2 INHIBITOR/HISTONE DEACETYLASE INHIBITOR COMBINATION |
| US2008213399 | 9-5-2008 | Combination Therapies Using Hdac Inhibitors |
| US2008194690 | 8-15-2008 | Pharmaceutical Formulations Of Hdac Inhibitors |
| US7407988 | 8-6-2008 | Carbamic acid compounds comprising a sulfonamide linkage as HDAC inhibitors |
| US7402603 | 7-23-2008 | Cyclooxygenase-2 inhibitor/histone deacetylase inhibitor combination |
| US7183298 | 2-28-2007 | Carbamic acid compounds comprising a sulfonamide linkage as HDAC inhibitors |
| US2005107445 | 5-20-2005 | Carbamic acid compounds comprising a sulfonamide linkage as HDAC inhibitors |
| US6888027 | 5-4-2005 | Carbamic acid compounds comprising a sulfonamide linkage as hdac inhibitors |
| WO2002030879A2 | Sep 27, 2001 | Apr 18, 2002 | Prolifix Ltd | Carbamic acid compounds comprising asulfonamide linkage as hdac inhibitors |
| US7973181 | 7-6-2011 | HYDROXAMIC ACID DERIVATIVES AS INHIBITORS OF HDAC ENZYMATIC ACTIVITY |
| US7928081 | 4-20-2011 | Combined Use of Prame Inhibitors and Hdac Inhibitors |
| US2011077305 | 3-32-2011 | 5-LIPOXYGENASE INHIBITORS |
| US2011003777 | 1-7-2011 | Methods of Treatment Employing Prolonged Continuous Infusion of Belinostat |
| US2010286279 | 11-12-2010 | Methods of Synthesis of Certain Hydroxamic Acid Compounds |
| US2010190694 | 7-30-2010 | Methods for identifying patients who will respond well to cancer treatment |
| US2010010010 | 1-15-2010 | HDAC INHIBITORS |
| US2009312311 | 12-18-2009 | COMBINATION OF ORGANIC COMPOUNDS |
| US2009192211 | 7-31-2009 | CYCLOOXYGENASE-2 INHIBITOR/HISTONE DEACETYLASE INHIBITOR COMBINATION |
| US7557140 | 7-8-2009 | CARBAMIC ACID COMPOUNDS COMPRISING A SULFONAMIDE LINKAGE AS HDAC INHIBITORS |
| WO1998038859A1 * | Mar 4, 1998 | Sep 11, 1998 | Thomas E Barta | Sulfonyl divalent aryl or heteroaryl hydroxamic acid compounds |
| WO1999024399A1 * | Nov 12, 1998 | May 20, 1999 | Darwin Discovery Ltd | Hydroxamic and carboxylic acid derivatives having mmp and tnf inhibitory activity |
| WO2000056704A1 * | Mar 22, 2000 | Sep 28, 2000 | Duncan Batty | Hydroxamic and carboxylic acid derivatives |
| WO2000069819A1 * | May 12, 2000 | Nov 23, 2000 | Thomas E Barta | Hydroxamic acid derivatives as matrix metalloprotease inhibitors |
| WO2001038322A1 * | Nov 22, 2000 | May 31, 2001 | Methylgene Inc | Inhibitors of histone deacetylase |
| EP0570594A1 * | Dec 7, 1992 | Nov 24, 1993 | SHIONOGI & CO., LTD. | Hydroxamic acid derivative based on aromatic sulfonamide |
| EP0931788A2 * | Dec 16, 1998 | Jul 28, 1999 | Pfizer Inc. | Metalloprotease inhibitors |
| GB2312674A * | Title not available |
| WO2002030879A2 | Sep 27, 2001 | Apr 18, 2002 | Prolifix Ltd | Carbamic acid compounds comprising a sulfonamide linkage as hdac inhibitors |
| WO2005063806A1 | Dec 30, 2003 | Jul 14, 2005 | Council Scient Ind Res | Arginine hydrochloride enhances chaperone-like activity of alpha crystallin |
| US4642316 | May 20, 1985 | Feb 10, 1987 | Warner-Lambert Company | Parenteral phenytoin preparations |
| WO2008090585A2 * | Jan 25, 2008 | Jul 31, 2008 | Univ Roma | Soluble forms of inclusion complexes of histone deacetylase inhibitors and cyclodextrins, their preparation processes and uses in the pharmaceutical field |
| WO2009109861A1 * | Mar 6, 2009 | Sep 11, 2009 | Topotarget A/S | Methods of treatment employing prolonged continuous infusion of belinostat |
| WO2010048332A2 * | Oct 21, 2009 | Apr 29, 2010 | Acucela, Inc. | Compounds for treating ophthalmic diseases and disorders |
| WO2011064663A1 | Nov 24, 2010 | Jun 3, 2011 | Festuccia, Claudio | Combination treatment employing belinostat and bicalutamide |
| US20110003777 * | Mar 6, 2009 | Jan 6, 2011 | Topotarget A/S | Methods of Treatment Employing Prolonged Continuous Infusion of Belinostat |
………………………..
SPECTRUM
Tiny Biotech With Three Cancer Drugs Is More Alluring Takeover Bet Now
Forbes
The drug is one of Spectrum’s two drugs undergoing phase 3 clinical trials. Allergan paid Spectrum $41.5 million and will make additional payments of up to $304 million based on achieving certain milestones. So far, Raj Shrotriya, Spectrum’s chairman, …
……………………………..

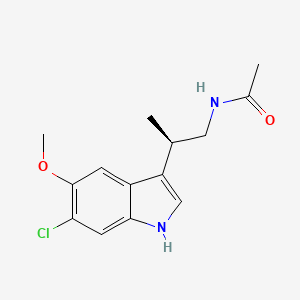
Panobinostat
HDAC inhibitors, orphan drug
cas 404950-80-7
2E)-N-hydroxy-3-[4-({[2-(2-methyl-1H-indol-3-yl)ethyl]amino}methyl)phenyl]acrylamide
N-hydroxy-3-[4-[[[2-(2-methyl-1H-indol-3-yl)ethyl]amino]methyl]phenyl]-2E-2-propenamide (alternatively, N-hydroxy-3-(4-{[2-(2-methyl-1H-indol-3-yl)-ethylamino]-methyl}-phenyl)-acrylamide)
Molecular Formula: C21H23N3O2 Molecular Weight: 349.42622
A hydroxamic acid analog histone deacetylase inhibitor from Novartis.
NOVARTIS, innovator
Histone deacetylase inhibitors
Is currently being examined in cutaneous T-cell lymphoma, CML and breast cancer.
clinical trials click here phase 3
DRUG SUBSTANCE–LACTATE AS IN http://www.google.com/patents/US7989639 SEE EG 31

Panobinostat (LBH-589) is an experimental drug developed by Novartis for the treatment of various cancers. It is a hydroxamic acid[1] and acts as a non-selective histone deacetylase inhibitor (HDAC inhibitor).[2]
panobinostat
Panobinostat is a cinnamic hydroxamic acid analogue with potential antineoplastic activity. Panobinostat selectively inhibits histone deacetylase (HDAC), inducing hyperacetylation of core histone proteins, which may result in modulation of cell cycle protein expression, cell cycle arrest in the G2/M phase and apoptosis. In addition, this agent appears to modulate the expression of angiogenesis-related genes, such as hypoxia-inducible factor-1alpha (HIF-1a) and vascular endothelial growth factor (VEGF), thus impairing endothelial cell chemotaxis and invasion. HDAC is an enzyme that deacetylates chromatin histone proteins. Check for
As of August 2012, it is being tested against Hodgkin’s Lymphoma, cutaneous T cell lymphoma (CTCL)[3] and other types of malignant disease in Phase III clinical trials, against myelodysplastic syndromes, breast cancer and prostate cancer in Phase II trials, and against chronic myelomonocytic leukemia (CMML) in a Phase I trial.[4][5]
Panobinostat is a histone deacetylase (HDAC) inhibitor which was filed for approval in the U.S. in 2010 for the oral treatment of relapsed/refractory classical Hodgkin’s lymphoma in adult patients. The company is conducting phase II/III clinical trials for the oral treatment of multiple myeloma, chronic myeloid leukemia and myelodysplasia. Phase II trials are also in progress for the treatment of primary myelofibrosis, post-polycythemia Vera, post-essential thrombocytopenia, Waldenstrom’s macroglobulinemia, recurrent glioblastoma (GBM) and for the treatment of pancreatic cancer progressing on gemcitabine therapy. Additional trials are under way for the treatment of hematological neoplasms, prostate cancer, colorectal cancer, renal cell carcinoma, non-small cell lung cancer (NSCLC), malignant mesothelioma, acute lymphoblastic leukemia, acute myeloid leukemia, head and neck cancer and gastrointestinal neuroendocrine tumors. Early clinical studies are also ongoing for the treatment of HER2 positive metastatic breast cancer. Additionally, phase II clinical trials are ongoing at Novartis as well as Neurological Surgery for the treatment of recurrent malignant gliomas as are phase I/II initiated for the treatment of acute graft versus host disease. The National Cancer Institute had been conducting early clinical trials for the treatment of metastatic hepatocellular carcinoma; however, these trials were terminated due to observed dose-limiting toxicity. In 2009, Novartis terminated its program to develop panobinostat for the treatment of cutaneous T-cell lymphoma. A program for the treatment of small cell lung cancer was terminated in 2012. Phase I clinical trials are ongoing for the treatment of metastatic and/or malignant melanoma and for the treatment of sickle cell anemia. The University of Virginia is conducting phase I clinical trials for the treatment of newly diagnosed and recurrent chordoma in combination with imatinib. Novartis is evaluating panobinostat for its potential to re-activate HIV transcription in latently infected CD4+ T-cells among HIV-infected patients on stable antiretroviral therapy.
Mechanistic evaluations revealed that panobinostat-mediated tumor suppression involved blocking cell-cycle progression and gene transcription induced by the interleukin IL-2 promoter, accompanied by an upregulation of p21, p53 and p57, and subsequent cell death resulted from the stimulation of caspase-dependent and -independent apoptotic pathways and an increase in the mitochondrial outer membrane permeability. In 2007, the compound received orphan drug designation in the U.S. for the treatment of cutaneous T-cell lymphoma and in 2009 and 2010, orphan drug designation was received in the U.S. and the E.U., respectively, for the treatment of Hodgkin’s lymphoma. This designation was also assigned in 2012 in the U.S. and the E.U. for the treatment of multiple myeloma.
Cardiovascular disease is the leading cause of morbidity and mortality in the western world and during the last decades it has also become a rapidly increasing problem in developing countries. An estimated 80 million American adults (one in three) have one or more expressions of cardiovascular disease (CVD) such as hypertension, coronary heart disease, heart failure, or stroke. Mortality data show that CVD was the underlying cause of death in 35% of all deaths in 2005 in the United States, with the majority related to myocardial infarction, stroke, or complications thereof. The vast majority of patients suffering acute cardiovascular events have prior exposure to at least one major risk factor such as cigarette smoking, abnormal blood lipid levels, hypertension, diabetes, abdominal obesity, and low-grade inflammation.
Pathophysiologically, the major events of myocardial infarction and ischemic stroke are caused by a sudden arrest of nutritive blood supply due to a blood clot formation within the lumen of the arterial blood vessel. In most cases, formation of the thrombus is precipitated by rupture of a vulnerable atherosclerotic plaque, which exposes chemical agents that activate platelets and the plasma coagulation system. The activated platelets form a platelet plug that is armed by coagulation-generated fibrin to form a biood clot that expands within the vessel lumen until it obstructs or blocks blood flow, which results in hypoxic tissue damage (so-called infarction). Thus, thrombotic cardiovascular events occur as a result of two distinct processes, i.e. a slowly progressing long-term vascular atherosclerosis of the vessel wall, on the one hand, and a sudden acute clot formation that rapidly causes flow arrest, on the other. This invention solely relates to the latter process.
Recently, inflammation has been recognized as an important risk factor for thrombotic events. Vascular inflammation is a characteristic feature of the atherosclerotic vessel wall, and inflammatory activity is a strong determinant of the susceptibility of the atherosclerotic plaque to rupture and initiate intravascular clotting. Also, autoimmune conditions with systemic inflammation, such as rheumatoid arthritis, systemic lupus erythematosus and different forms of vasculitides, markedly increase the risk of myocardial infarction and stroke.
Traditional approaches to prevent and treat cardiovascular events are either targeted 1) to slow down the progression of the underlying atherosclerotic process, 2) to prevent clot formation in case of a plaque rupture, or 3) to direct removal of an acute thrombotic flow obstruction. In brief, antiatherosclerotic treatment aims at modulating the impact of general risk factors and includes dietary recommendations, weight loss, physical exercise, smoking cessation, cholesterol- and blood pressure treatment etc. Prevention of clot formation mainly relies on the use of antiplatelet drugs that inhibit platelet activation and/or aggregation, but also in some cases includes thromboembolic prevention with oral anticoagulants such as warfarin. Post-hoc treatment of acute atherothrombotic events requires either direct pharmacological lysis of the clot by thrombolytic agents such as recombinant tissue-type plasminogen activator or percutaneous mechanical dilation of the obstructed vessel.
Despite the fact that multiple-target antiatherosclerotic therapy and clot prevention by antiplatelet agents have lowered the incidence of myocardial infarction and ischemic stroke, such events still remain a major population health problem. This shows that in patients with cardiovascular risk factors these prophylactic measures are insufficient to completely prevent the occurrence of atherothrombotic events.
Likewise, thrombotic conditions on the venous side of the circulation, as well as embolic complications thereof such as pulmonary embolism, still cause substantial morbidity and mortality. Venous thrombosis has a different clinical presentation and the relative importance of platelet activation versus plasma coagulation are somewhat different with an preponderance for the latter in venous thrombosis, However, despite these differences, the major underlying mechanisms that cause thrombotic vessel occlusions are similar to those operating on the arterial circulation. Although unrelated to atherosclerosis as such, the risk of venous thrombosis is related to general cardiovascular risk factors such as inflammation and metabolic aberrations.

Panobinostat can be synthesized as follows: Reduction of 2-methylindole-3-glyoxylamide (I) with LiAlH4 affords 2-methyltryptamine (II). 4-Formylcinnamic acid (III) is esterified with methanolic HCl, and the resulting aldehyde ester (IV) is reductively aminated with 2-methyltryptamine (II) in the presence of NaBH3CN (1) or NaBH4 (2) to give (V). The title hydroxamic acid is then obtained by treatment of ester (V) with aqueous hydroxylamine under basic conditions.
Panobinostat is currently being used in a Phase I/II clinical trial that aims at curing AIDS in patients on highly active antiretroviral therapy (HAART). In this technique panobinostat is used to drive the HI virus’s DNA out of the patient’s DNA, in the expectation that the patient’s immune system in combination with HAART will destroy it.[6][7]
Panobinostat has been found to synergistically act with sirolimus to kill pancreatic cancer cells in the laboratory in a Mayo Clinic study. In the study, investigators found that this combination destroyed up to 65 percent of cultured pancreatic tumor cells. The finding is significant because the three cell lines studied were all resistant to the effects of chemotherapy – as are many pancreatic tumors.[8]
Panobinostat has also been found to significantly increase in vitro the survival of motor neuron (SMN) protein levels in cells of patients suffering fromspinal muscular atrophy.[9]
Panobinostat was able to selectively target triple negative breast cancer (TNBC) cells by inducing hyperacetylation and cell cycle arrest at the G2-M DNA damage checkpoint; partially reversing the morphological changes characteristic of breast cancer cells.[10]
Panobinostat, along with other HDAC inhibitors, is also being studied for potential to induce virus HIV-1 expression in latently infected cells and disrupt latency. These resting cells are not recognized by the immune system as harboring the virus and do not respond to antiretroviral drugs.[11]
Panobinostat inhibits multiple histone deacetylase enzymes, a mechanism leading to apoptosis of malignant cells via multiple pathways.[1]
The compound N-hydroxy-3-[4-[[[2-(2-methyl-1H-indol-3-yl)ethyl]amino]methyl]phenyl]-2E-2-propenamide (alternatively, N-hydroxy-3-(4-{[2-(2-methyl-1H-indol-3-yl)-ethylamino]-methyl}-phenyl)-acrylamide) has the formula
as described in WO 02/22577. Valuable pharmacological properties are attributed to this compound; thus, it can be used, for example, as a histone deacetylase inhibitor useful in therapy for diseases which respond to inhibition of histone deacetylase activity. WO 02/22577 does not disclose any specific salts or salt hydrates or solvates of N-hydroxy-3-[4-[[[2-(2-methyl-1H-indol-3-yl)ethyl]amino]methyl]phenyl]-2E-2-propenamide.
The compounds described above are often used in the form of a pharmaceutically acceptable salt. Pharmaceutically acceptable salts include, when appropriate, pharmaceutically acceptable base addition salts and acid addition salts, for example, metal salts, such as alkali and alkaline earth metal salts, ammonium salts, organic amine addition salts, and amino acid addition salts, and sulfonate salts. Acid addition salts include inorganic acid addition salts such as hydrochloride, sulfate and phosphate, and organic acid addition salts such as alkyl sulfonate, arylsulfonate, acetate, maleate, fumarate, tartrate, citrate and lactate. Examples of metal salts are alkali metal salts, such as lithium salt, sodium salt and potassium salt, alkaline earth metal salts such as magnesium salt and calcium salt, aluminum salt, and zinc salt. Examples of ammonium salts are ammonium salt and tetramethylammonium salt. Examples of organic amine addition salts are salts with morpholine and piperidine. Examples of amino acid addition salts are salts with glycine, phenylalanine, glutamic acid and lysine. Sulfonate salts include mesylate, tosylate and benzene sulfonic acid salts.
……………………………..
GENERAL METHOD OF SYNTHESIS
ADD YOUR METHYL AT RIGHT PLACE
As is evident to those skilled in the art, the many of the deacetylase inhibitor compounds of the present invention contain asymmetric carbon atoms. It should be understood, therefore, that the individual stereoisomers are contemplated as being included within the scope of this invention.
The hydroxamate compounds of the present invention can be produced by known organic synthesis methods. For example, the hydroxamate compounds can be produced by reacting methyl 4-formyl cinnamate with tryptamine and then converting the reactant to the hydroxamate compounds. As an example, methyl 4-formyl cinnamate 2, is prepared by acid catalyzed esterification of 4-formylcinnamic acid 3 (Bull. Chem. Soc. Jpn. 1995; 68:2355-2362). An alternate preparation of methyl 4-formyl cinnamate 2 is by a Pd- catalyzed coupling of methyl acrylate 4 with 4-bromobenzaldehyde 5.
CHO
Additional starting materials can be prepared from 4-carboxybenzaldehyde 6, and an exemplary method is illustrated for the preparation of aldehyde 9, shown below. The carboxylic acid in 4-carboxybenzaldehyde 6 can be protected as a silyl ester (e.g., the t- butyldimethylsilyl ester) by treatment with a silyl chloride (e.g., f-butyldimethylsilyl chloride) and a base (e.g. triethylamine) in an appropriate solvent (e.g., dichloromethane). The resulting silyl ester 7 can undergo an olefination reaction (e.g., a Horner-Emmons olefination) with a phosphonate ester (e.g., triethyl 2-phosphonopropionate) in the presence of a base (e.g., sodium hydride) in an appropriate solvent (e.g., tetrahydrofuran (THF)). Treatment of the resulting diester with acid (e.g., aqueous hydrochloric acid) results in the hydrolysis of the silyl ester providing acid 8. Selective reduction of the carboxylic acid of 8 using, for example, borane-dimethylsuflide complex in a solvent (e.g., THF) provides an intermediate alcohol. This intermediate alcohol could be oxidized to aldehyde 9 by a number of known methods, including, but not limited to, Swern oxidation, Dess-Martin periodinane oxidation, Moffatt oxidation and the like.
The aldehyde starting materials 2 or 9 can be reductively aminated to provide secondary or tertiary amines. This is illustrated by the reaction of methyl 4-formyl cinnamate 2 with tryptamine 10 using sodium triacetoxyborohydride (NaBH(OAc)3) as the reducing agent in dichloroethane (DCE) as solvent to provide amine 11. Other reducing agents can be used, e.g., sodium borohydride (NaBH ) and sodium cyanoborohydride (NaBH3CN), in other solvents or solvent mixtures in the presence or absence of acid catalysts (e.g., acetic acid and trifluoroacetic acid). Amine 11 can be converted directly to hydroxamic acid 12 by treatment with 50% aqueous hydroxylamine in a suitable solvent (e.g., THF in the presence of a base, e.g., NaOH). Other methods of hydroxamate formation are known and include reaction of an ester with hydroxylamine hydrochloride and a base (e.g., sodium hydroxide or sodium methoxide) in a suitable solvent or solvent mixture (e.g., methanol, ethanol or methanol/THF).
NOTE ….METHYL SUBSTITUENT ON 10 WILL GIVE YOU PANOBINOSTAT

……………………………….
Journal of Medicinal Chemistry, 2011 , vol. 54, 13 pg. 4694 – 4720
(E)-N-Hydroxy-3-(4-{[2-(2-methyl-1H-indol-3-yl)-ethylamino]-methyl}-phenyl)-acrylamide
lactate
(34, panobinostat, LBH589)
http://pubs.acs.org/doi/full/10.1021/jm2003552
http://pubs.acs.org/doi/suppl/10.1021/jm2003552/suppl_file/jm2003552_si_001.pdf
for str see above link
α-methyl-β-(β-bromoethyl)indole (29) was made according to method reported by Grandberg et al.(2. Grandberg, I. I.; Kost, A. N.; Terent’ev, A. P. Reactions of hydrazine derivatives. XVII. New synthesis of α-methyltryptophol. Zhurnal Obshchei Khimii 1957, 27, 3342–3345. )
The bromide 29 was converted to amine 30 by using similar method used by Sletzinger et al.(3. Sletzinger, M.; Ruyle, W. V.; Waiter, A. G. (Merck & Co., Inc.). Preparation of tryptamine
derivatives. U.S. Patent US 2,995,566, Aug 8, 1961.)
To a 500 mL flask, crude 2-methyltryptamine 30 (HPLC purity 75%, 1.74 g, 7.29 mmol) and 3-(4-
formyl-phenyl)-acrylic acid methyl ester 31 (HPLC purity 84%, 1.65 g, 7.28 mmol) were added,
followed by DCM (100 mL) and MeOH (30 mL). The clear solution was stirred at room temp for 30
min, then NaBH3CN (0.439 g, 6.99 mmol) was added in small portions. The reaction mixture was
stirred at room temp overnight. After removal of the solvents, the residue was diluted with DCM and
added saturated NaHCO3 aqueous solution, extracted with DCM twice. The DCM layer was dried
and concentrated, and the resulting residue was purified by flash chromatography (silica, 0–10%
MeOH in DCM) to afford 33 as orange solid (1.52 g, 60%). LC–MS m/z 349.2 ([M + H]+). 33 was
converted to hydroxamic acid 34 according to procedure D (Experimental Section), and the freebase
34 was treated with 1 equiv of lactic acid in MeOH–water (7:3) to form lactic acid salt which was
further recrystallized in MeOH–EtOAc to afford the lactic acid salt of 34as pale yellow solid. LC–MS m/z 350.2 ([M + H − lactate]+).
= DELTA
1H NMR (DMSO-d6) 10.72 (s, 1H, NH), 7.54 (d, J = 8.0 Hz, 2H), 7.44 (d, J = 16 Hz, 1H), 7.43 (d, J = 7.8 Hz, 2H), 7.38 (d, J = 7.6 Hz, 1H), 7.22 (d, J = 7.8 Hz, 1H), 6.97 (td, J = 7.8 Hz, 1H), 7.44 (d, J = 15.8 Hz, 1H), 7.22 (t, J = 7.8 Hz, 2H), 7.08 (d, J = 7.8Hz, 2H), 7.01 (t, J = 7.4, 0.9 Hz, 1H), 6.91 (td, J = 7.4, 0.9 Hz, 1H), 6.47 (d, J = 15.2 Hz, 1H), 3.94(q, J = 6.8 Hz, 1H, lactate CH), 3.92 (s, 2H), 2.88 and 2.81 (m, each, 4H, AB system, CH2CH2),2.31 (s, 3H), 1.21 (d, J = 6.8 Hz, 3H).;
13C NMR (DMSO-d6) 176.7 (lactate C=O), 162.7, 139.0,
137.9, 135.2, 134.0, 132.1, 129.1, 128.1, 127.4, 119.9, 119.0, 118.1, 117.2, 110.4, 107.0, 66.0, 51.3,
48.5, 22.9, 20.7, 11.2.
…………………………………………..
PANOBINOSTAT DRUG SUBSTANCE SYNTHESIS AND DATA
http://www.google.com/patents/US7989639

A flow diagram for the synthesis of LBH589 lactate is provided in FIG. A. A nomenclature reference index of the intermediates is provided below in the Nomenclature Reference Index:
| Nomenclature reference index | |
| Compound | Chemical name |
| 1 | 4-Bromo-benzaldehyde |
| 2 | Methyl acrylate |
| 3 | (2E)-3-(formylphenyl)-2-propenoic acid, methyl ester |
| 4 | 3-[4-[[[2-(2-Methyl-1H-indol-3- |
| yl)ethyl]amino]methyl]phenyl]-2- | |
| propenoic acid, methyl ester, monohydrochloride | |
| 5 | (2E)-N-hydroxy-3-[4-[[[2-(2-methyl-1H-indol-3- |
| yl)ethyl]amino]methyl]phenyl]-2-propenamide | |
| 6 | 2-hydroxypropanoic acid, compd. with 2(E)-N- |
| hydroxy-3-[4-[[[2-(2-methyl-1H- | |
| indol-3-yl)ethyl]amino]methyl]phenyl]-2-propenamide | |
| Z3a | 2-Methyl-1H-indole-3-ethanamine |
| Z3b | 5-Chloro-2-pentanone |
| Z3c | Phenylhydrazine |
The manufacture of LBH589 lactate (6) drug substance is via a convergent synthesis; the point of convergence is the condensation of indole-amine Z3a with aldehyde 3.
The synthesis of indole-amine Z3a involves reaction of 5-chloro-2 pentanone (Z3b) with phenylhydrazine (Z3c) in ethanol at reflux (variation of Fischer indole synthesis).
Product isolation is by an extractive work-up followed by crystallization. Preparation of aldehyde 3 is by palladium catalyzed vinylation (Heck-type reaction; Pd(OAc)2/P(o-Tol)3/Bu3N in refluxing CH3CN) of 4-bromo-benzyladehyde (1) with methyl acrylate (2) with product isolation via precipitation from dilute HCl solution. Intermediates Z3a and 3 are then condensed to an imine intermediate, which is reduced using sodium borohydride in methanol below 0° C. (reductive amination). The product indole-ester 4, isolated by precipitation from dilute HCl, is recrystallized from methanol/water, if necessary. The indole ester 4 is converted to crude LBH589 free base 5 via reaction with hydroxylamine and sodium hydroxide in water/methanol below 0° C. The crude LBH589 free base 5 is then purified by recrystallization from hot ethanol/water, if necessary. LBH589 free base 5 is treated with 85% aqueous racemic lactic acid and water at ambient temperature. After seeding, the mixture is heated to approximately 65° C., stirred at this temperature and slowly cooled to 45-50° C. The resulting slurry is filtered and washed with water and dried to afford LBH589 lactate (6).
If necessary the LBH589 lactate 6 may be recrystallised once again from water in the presence of 30 mol % racemic lactic acid. Finally the LBH589 lactate is delumped to give the drug substance. If a rework of the LBH589 lactate drug substance 6 is required, the LBH589 lactate salt is treated with sodium hydroxide in ethanol/water to liberate the LBH589 free base 5 followed by lactate salt formation and delumping as described above.
All starting materials, reagents and solvents used in the synthesis of LBH589 lactate are tested according to internal specifications or are purchased from established suppliers against a certificate of analysis.
EXAMPLE 7 Formation of Monohydrate Lactate Salt
About 40 to 50 mg of N-hydroxy-3-[4-[[[2-(2-methyl-1H-indol-3-yl)ethyl]amino]methyl]phenyl]-2E-2-propenamide free base was suspended in 1 ml of a solvent as listed in Table 7. A stoichiometric amount of lactic acid was subsequently added to the suspension. The mixture was stirred at ambient temperature and when a clear solution formed, stirring continued at 4° C. Solids were collected by filtration and analyzed by XRPD, TGA and 1H-NMR.
| TABLE 7 | |||||
| LOD, % | |||||
| Physical | Crystallinity | (Tdesolvation) | |||
| Solvent | T, ° C. | Appear. | and Form | Tdecomposit. | 1H-NMR |
| IPA | 4 | FFP | excellent | 4.3 (79.3) | — |
| HA | 156.3 | ||||
| Acetone | 4 | FFP | excellent | 4.5 (77.8) | 4.18 (Hbz) |
| HA | 149.5 | ||||
The salt forming reaction in isopropyl alcohol and acetone at 4° C. produced a stoichiometric (1:1) lactate salt, a monohydrate. The salt is crystalline, begins to dehydrate above 77° C., and decomposes above 150° C.
EXAMPLE 18 Formation of Anhydrous Lactate Salt
DL-lactic acid (4.0 g, 85% solution in water, corresponding to 3.4 g pure DL-lactic acid) is diluted with water (27.2 g), and the solution is heated to 90° C. (inner temperature) for 15 hours. The solution is allowed to cool down to room temperature and is used as lactic acid solution for the following salt formation step.
N-hydroxy-3-[4-[[[2-(2-methyl-1H-indol-3-yl)ethyl]amino]methyl]phenyl]-2E-2-propenamide free base (10.0 g) is placed in a 4-necked reaction flask with mechanical stirrer. Demineralized water (110.5 g) is added, and the suspension is heated to 65° C. (inner temperature) within 30 minutes. The DL-lactic acid solution is added to this suspension during 30 min at 65° C. During the addition of the lactate salt solution, the suspension converted into a solution. The addition funnel is rinsed with demineralized water (9.1 g), and the solution is stirred at 65° C. for an additional 30 minutes. The solution is cooled down to 45° C. (inner temperature) and seed crystals (10 mg N-hydroxy-3-[4-[[[2-(2-methyl-1H-indol-3-yl)ethyl]amino]methyl]phenyl]-2E-2-propenamide lactate monohydrate) are added at this temperature. The suspension is cooled down to 33° C. and is stirred for additional 20 hours at this temperature. The suspension is re-heated to 65° C., stirred for 1 hour at this temperature and is cooled to 33° C. within 1 hour. After additional stirring for 3 hours at 33° C., the product is isolated by filtration, and the filter cake is washed with demineralized water (2×20 g). The wet filter-cake is dried in vacuo at 50° C. to obtain the anhydrous N-hydroxy-3-[4-[[[2-(2-methyl-1H-indol-3-yl)ethyl]amino]methyl]phenyl]-2E-2-propenamide lactate salt as a crystalline product. The product is identical to the monohydrate salt (form HA) in HPLC and in 1H-NMR, with the exception of the integrals of water signals in the 1H-NMR spectra.
In additional salt formation experiments carried out according to the procedure described above, the product solution was filtered at 65° C. before cooling to 45° C., seeding and crystallization. In all cases, form A (anhydrate form) was obtained as product.
EXAMPLE 19 Formation of Anhydrous Lactate Salt
DL-lactic acid (2.0 g, 85% solution in water, corresponding to 1.7 g pure DL-lactic acid) is diluted with water (13.6 g), and the solution is heated to 90° C. (inner temperature) for 15 hours. The solution was allowed to cool down to room temperature and is used as lactic acid solution for the following salt formation step.
N-hydroxy-3-[4-[[[2-(2-methyl-1H-indol-3-yl)ethyl]amino]methyl]phenyl]-2E-2-propenamide free base (5.0 g) is placed in a 4-necked reaction flask with mechanical stirrer. Demineralized water (54.85 g) is added, and the suspension is heated to 48° C. (inner temperature) within 30 minutes. The DL-lactic acid solution is added to this suspension during 30 minutes at 48° C. A solution is formed. Seed crystals are added (as a suspension of 5 mg N-hydroxy-3-[4-[[[2-(2-methyl-1H-indol-3-yl)ethyl]amino]methyl]phenyl]-2E-2-propenamide lactate salt, anhydrate form A, in 0.25 g of water) and stirring is continued for 2 additional hours at 48° C. The temperature is raised to 65° C. (inner temperature) within 30 minutes, and the suspension is stirred for additional 2.5 hours at this temperature. Then the temperature is cooled down to 48° C. within 2 hours, and stirring is continued at this temperature for additional 22 hours. The product is isolated by filtration and the filter cake is washed with demineralized water (2×10 g). The wet filter-cake is dried in vacuo at 50° C. to obtain anhydrous N-hydroxy-3-[4-[[[2-(2-methyl-1H-indol-3-yl)ethyl]amino]methyl]phenyl]-2E-2-propenamide lactate salt (form A) as a crystalline product.
EXAMPLE 20 Conversion of Monohydrate Lactate Salt to Anhydrous Lactate Salt
DL-lactic acid (0.59 g, 85% solution in water, corresponding to 0.5 g pure DL-lactic acid) is diluted with water (4.1 g), and the solution is heated to 90° C. (inner temperature) for 15 hours. The solution is allowed to cool down to room temperature and is used as lactic acid solution for the following salt formation step.
10 g of N-hydroxy-3-[4-[[[2-(2-methyl-1H-indol-3-yl)ethyl]amino]methyl]phenyl]-2E-2-propenamide lactate salt monohydrate is placed in a 4-necked reaction flask. Water (110.9 g) is added, followed by the addition of the lactic acid solution. The addition funnel of the lactic acid is rinsed with water (15.65 g). The suspension is heated to 82° C. (inner temperature) to obtain a solution. The solution is stirred for 15 minutes at 82° C. and is hot filtered into another reaction flask to obtain a clear solution. The temperature is cooled down to 50° C., and seed crystals are added (as a suspension of 10 mg N-hydroxy-3-[4-[[[2-(2-methyl-1H-indol-3-yl)ethyl]amino]methyl]phenyl]-2E-2-propenamide lactate salt, anhydrate form, in 0.5 g of water). The temperature is cooled down to 33° C. and stirring is continued for additional 19 hours at this temperature. The formed suspension is heated again to 65° C. (inner temperature) within 45 minutes, stirred at 65° C. for 1 hour and cooled down to 33° C. within 1 hour. After stirring at 33° C. for additional 3 hours, the product is isolated by filtration and the wet filter cake is washed with water (50 g). The product is dried in vacuo at 50° C. to obtain crystalline anhydrous N-hydroxy-3-[4-[[[2-(2-methyl-1H-indol-3-yl) ethyl]amino]methyl]phenyl]-2E-2-propenamide lactate salt (form A).
EXAMPLE 21 Formation of Anhydrous Lactate Salt
DL-lactic acid (8.0 g, 85% solution in water, corresponding to 6.8 g pure DL-lactic acid) was diluted with water (54.4 g), and the solution was heated to 90° C. (inner temperature) for 15 hours. The solution was allowed to cool down to room temperature and was used as lactic acid solution for the following salt formation step.
N-hydroxy-3-[4-[[[2-(2-methyl-1H-indol-3-yl)ethyl]amino]methyl]phenyl]-2E-2-propenamide (20 g) is placed in a 1 L glass reactor, and ethanol/water (209.4 g of a 1:1 w/w mixture) is added. The light yellow suspension is heated to 60° C. (inner temperature) within 30 minutes, and the lactic acid solution is added during 30 minutes at this temperature. The addition funnel is rinsed with water (10 g). The solution is cooled to 38° C. within 2 hours, and seed crystals (20 mg of N-hydroxy-3-[4-[[[2-(2-methyl-1H-indol-3-yl)ethyl]amino]methyl]phenyl]-2E-2-propenamide lactate salt, anhydrate form) are added at 38° C. After stirring at 38° C. for additional 2 hours, the mixture is cooled down to 25° C. within 6 hours. Cooling is continued from 25° C. to 10° C. within 5 hours, from 10° C. to 5° C. within 4 hours and from 5° C. to 2° C. within 1 hour. The suspension is stirred for additional 2 hours at 2° C., and the product is isolated by filtration. The wet filter cake is washed with water (2×30 g), and the product is dried in vacuo at 45° C. to obtain crystalline anhydrous N-hydroxy-3-[4-[[[2-(2-methyl-1H-indol-3-yl)ethyl]amino]methyl]phenyl]-2E-2-propenamide lactate salt (form A).
EXAMPLE 28 Formation of Lactate Monohydrate Salt
3.67 g (10 mmol) of the free base monohydrate (N-hydroxy-3-[4-[[[2-(2-methyl-1H-indol-3-yl) ethyl]amino]methyl]phenyl]-2E-2-propenamide) and 75 ml of acetone were charged in a 250 ml 3-neck flask equipped with a magnetic stirrer and an addition funnel. To the stirred suspension were added dropwise 10 ml of 1 M lactic acid in water (10 mmol) dissolved in 20 ml acetone, affording a clear solution. Stirring continued at ambient and a white solid precipitated out after approximately 1 hour. The mixture was cooled in an ice bath and stirred for an additional hour. The white solid was recovered by filtration and washed once with cold acetone (15 ml). It was subsequently dried under vacuum to yield 3.94 g of the lactate monohydrate salt of N-hydroxy-3-[4-[[[2-(2-methyl-1H-indol-3-yl)ethyl]amino]methyl]phenyl]-2E-2-propenamide (86.2%).
| 23009203 | 11-26-2012 | Selective histone deacetylase 6 inhibitors bearing substituted urea linkers inhibit melanoma cell growth. | Journal of medicinal chemistry |
| 21634430 | 7-14-2011 | Discovery of (2E)-3-{2-butyl-1-[2-(diethylamino)ethyl]-1H-benzimidazol-5-yl}-N-hydroxyacrylamide (SB939), an orally active histone deacetylase inhibitor with a superior preclinical profile. | Journal of medicinal chemistry |
| 21417419 | 4-28-2011 | Discovery, synthesis, and pharmacological evaluation of spiropiperidine hydroxamic acid based derivatives as structurally novel histone deacetylase (HDAC) inhibitors. | Journal of medicinal chemistry |
| 19317450 | 4-23-2009 | Identification and characterization of small molecule inhibitors of a class I histone deacetylase from Plasmodium falciparum. | Journal of medicinal chemistry |
| 15650931 | 1-1-2005 | The American Society of Hematology–46th Annual Meeting and Exposition. HDAC, Flt and farnesyl transferase inhibitors. | IDrugs : the investigational drugs journal |
| US7989639 | 8-3-2011 | PROCESS FOR MAKING SALTS OF N-HYDROXY-3-[4-[[[2-(2-METHYL-1H-INDOL-3-YL)ETHYL]AMINO]METHYL]PHENYL]-2E-2-PROPENAMIDE |
| US2010286409 | 11-12-2010 | SALTS OF N-HYDROXY-3-[4-[[[2-(2-METHYL-1H-INDOL-3-YL)ETHYL]AMINO]METHYL]PHENYL]-2E-2-PROPENAMIDE |
| US2010179208 | 7-16-2010 | Use of HDAC Inhibitors for the Treatment of Bone Destruction |
| US2010160257 | 6-25-2010 | USE OF HDAC INHIBITORS FOR THE TREATMENT OF MYELOMA |
| US2010137398 | 6-4-2010 | USE OF HDAC INHIBITORS FOR THE TREATMENT OF GASTROINTESTINAL CANCERS |
| US2009306405 | 12-11-2009 | PROCESS FOR MAKING N-HYDROXY-3-[4-[[[2-(2-METHYL-1H-INDOL-3-YL)ETHYL]AMINO]METHYL]PHENYL]-2E-2-PROPENAMIDE AND STARTING MATERIALS THEREFOR |
| US2009281159 | 11-13-2009 | USE OF HDAC INHIBITORS FOR THE TREATMENT OF LYMPHOMAS |
| US2009264439 | 10-23-2009 | Combination of a) N–4-(3-pyridyl)-2-pyrimidine-amine and b) a histone deacetylase inhibitor for the treatment of leukemia |
| US2009197936 | 8-7-2009 | SALTS OF N-HYDROXY-3-[4-[[[2-(2-METHYL-1H-INDOL-3-YL)ETHYL]AMINO]METHYL]PHENYL]-2E-2-PROPENAMIDE |
| US2009012066 | 1-9-2009 | Method of Use of Deacetylase Inhibitors |
| US2008319045 | 12-26-2008 | Combination of Histone Deacetylase Inhibitors and Radiation |
| US2008221126 | 9-12-2008 | Use of Hdac Inhibitors for the Treatment of Myeloma |
| US2008176849 | 7-25-2008 | DEACETYLASE INHIBITORS |
| US2006189674 | 8-25-2006 | Deacetylase inhibitors |
| US7067551 | 6-28-2006 | Deacetylase inhibitors |
| US2006100140 | 5-12-2006 | Combination of a) n-{5-[4-(4-methyl-piperazino-methyl)-benzoylamido]2-methylphenyl}-4- (3-pyridyl)-2-pyrimidine-amine and b) a histone deacetylase inhibitor for the treatment of leukemia |
| US6833384 | 12-22-2004 | Deacetylase inhibitors |
| US6552065 | 4-23-2003 | Deacetylase inhibitors |
| GB776693A | Title not available | |||
| GB891413A | Title not available | |||
| GB2185020A | Title not available | |||
| WO2002022577A2 | Aug 30, 2001 | Mar 21, 2002 | Kenneth Walter Bair | Hydroxamate derivatives useful as deacetylase inhibitors |
| WO2003016307A1 | Aug 6, 2002 | Aug 19, 1993 | Jolie Anne Bastian | β3 ADRENERGIC AGONISTS |
| WO2003039599A1 | Nov 5, 2002 | May 15, 2003 | Ying-Nan Pan Chen | Cyclooxygenase-2 inhibitor/histone deacetylase inhibitor combination |
| WO2005105740A2 | Apr 26, 2005 | Nov 10, 2005 | Serguei Fine | Preparation of tegaserod and tegaserod maleate |
| WO2006021397A1 | Aug 22, 2005 | Mar 2, 2006 | Recordati Ireland Ltd | Lercanidipine salts |
…………………………………..
extras
5. Mocetinostat (MGCD0103), including pharmaceutically acceptable salts thereof. Balasubramanian et al., Cancer Letters 280: 211-221 (2009).
Mocetinostat, has the following chemical structure and name:
Vorinostat, including pharmaceutically acceptable salts thereof. Marks et al., Nature Biotechnology 25, 84 to 90 (2007); Stenger, Community Oncology 4, 384-386 (2007).
Vorinostat has the following chemical structure and name:
Belinostat (PXD-101 , PX-105684)
(2E)-3-[3-(anilinosulfonyl)phenyl]-N-hydroxyacrylamide
……………………………………………….
Dacinostat (LAQ-824, NVP-LAQ824,)
((E)-N-hydroxy-3-[4-[[2-hydroxyethyl-[2-(1 H-indol-3-yl)ethyl]amino]methyl]phenyl]prop-2-enamide
Entinostat (MS-275, SNDX-275, MS-27-275)
4-(2-aminophenylcarbamoyl)benzylcarbamate
(a) The HDAC inhibitor Vorinostat™ or a salt, hydrate, or solvate thereof.
Vorinostat………………..
(b) The HDAC inhibitor Givinostat or a salt, hydrate, or solvate thereof.
Givinostat or a salt, hydrate, or solvate thereof.

TASIMELTION, an orphan drug for non24
N-([(1R,2R)-2-(2,3-Dihydro-1-benzofuran-4-yl)cyclopropyl]methyl)propanamide
(1R-trans)-N-[[2-(2,3-dihydro-4-benzofuranyl)cyclopropyl]methyl]pro- pananamide VEC162
(-)-(trans)-N-[[2-(2,3-Dihydrobenzofuran-4-yl)cycloprop-1-yl]methyl]propanamide
N-(((1R,2R)-2-(2,3-Dihydro-1-benzofuran-4-yl)cyclopropyl)methyl)propanamide
Bristol-Myers Squibb Company
PRODUCT PATENT
U.S. Pat. No. 5,856,529
| CAS number | 609799-22-6 |
|---|
| Formula | C15H19NO2 |
|---|---|
| Mol. mass | 245.3 g/mol |
VEC-162, BMS-214778, 609799-22-6, Hetlioz, UNII-SHS4PU80D9,
January 31, 2014 — The U.S. Food and Drug Administration today approved Hetlioz (tasimelteon), a melatonin receptor agonist, to treat non-24- hour sleep-wake disorder (“non-24”) in totally blind individuals. Non-24 is a chronic circadian rhythm (body clock) disorder in the blind that causes problems with the timing of sleep. This is the first FDA approval of a treatment for the disorder.
Non-24 occurs in persons who are completely blind. Light does not enter their eyes and they cannot synchronize their body clock to the 24-hour light-dark cycle.
Tasimelteon
A year-long (2011-2012) study at Harvard is testing the use of tasimelteon in blind subjects with non-24-hour sleep–wake disorder.[4] In May 2013Vanda Pharmaceuticals submitted a New Drug Application to the Food and Drug Administration for Tasimelteon for the treatment of non-24-hour sleep–wake disorder in totally blind people.[5]
SEQUENCE
Discovered by Bristol-Myers Squibb (BMS) and co-developed with Vanda Pharmaceuticals, tasimelteon is a hypnotic family benzofuran. In Phase III development, it has an orphan drug status.
JAN2014.. APPROVED FDA
In mid-November 2013 the FDA announced their recommendation for the approval of Tasimelteon for the treatment of non-24-disorder.Tasimelteon effectively resets the circadian rhythm, helping to restore normal sleep patterns.http://www.fda.gov/downloads/AdvisoryCommittees/CommitteesMeetingMaterials/Drugs/PeripheralandCentralNervousSystemDrugsAdvisoryCommittee/UCM374388.pdf
January 2010: FDA granted orphan drug tasimelteon to disturbed sleep / wake in blind without light perception.
February 2008: Vanda has completed enrollment in its Phase III trial in chronic primary insomnia.
June 2007: Results of a Phase III trial for transient insomnia tasimelteon presented by Vanda at the 21st annual meeting of the Associated Professional Sleep Societies. These results demonstrated improvements in objective and subjective measures of sleep and its maintenance.
2004 Vanda gets a license tasimelteon (or BMS-214778 and VEC-162) from Bristol-Myers Squibb.
About Tasimelteon: Tasimelteon is a circadian regulator in development for the treatment of Non-24. Tasimelteon is a dual melatonin receptor agonist (DMRA) with selective agonist activityat the MT1 and MT2 receptors.Tasimelteon’s ability to reset the master body clock in the suprachiasmatic nucleus (SCN) results in the entrainment of the body’s melatonin and cortisol rhythms with the 24-hour day-night cycle. The patent claiming tasimelteon as a new chemical entity extends through December 2022, assuming a 5-year extension to be granted under the Hatch-Waxman Act. Tasimelteon has been granted orphan drug designation for the treatment of Non-24 from both the U.S. and the European Union.
Previously, BMS-214778, identified as an agonist of melatonin receptors, has been the subject of pre-clinical studies for the treatment of sleep disorders resulting from a disturbance of circadian rhythms.The first Pharmacokinetic studies were performed in rats and monkeys.
The master body clock controls the timing of many aspects of physiology, behavior and metabolism that show daily rhythms, including the sleep-wake cycles, body temperature, alertness and performance, metabolic rhythms and certain hormones which exhibit circadian variation. Outputs from the suprachiasmatic nucleus (SCN) control many endocrine rhythms including those of melatonin secretion by the pineal gland as well as the control of cortisol secretion via effects on the hypothalamus, the pituitary and the adrenal glands.
This master body clock, located in the SCN, spontaneously generates rhythms of approximately 24.5 hours. These non-24-hour rhythms are synchronized each day to the 24-hour day-night cycle by light, the primary environmental time cue which is detected by specialized cells in the retina and transmitted to the SCN via the retino-hypothalamic tract. Inability to detect this light signal, as occurs in most totally blind individuals, leads to the inability of the master body clock to be reset daily and maintain entrainment to a 24-hour day.
Non-24-Hour Disorder
Non-24, also referred to as Non-24-Hour Sleep-Wake Disorder (N24HSWD) or Non-24-Hour Disorder, is an orphan indication affecting approximately 65,000 to 95,000 people in the U.S. and 140,000 in Europe. Non-24 occurs when individuals, primarily blind with no light perception, are unable to synchronize their endogenous circadian pacemaker to the 24-hour light/dark cycle. Without light as a synchronizer, and because the period of the internal clock is typically a little longer than 24 hours, individuals with Non-24 experience their circadian drive to initiate sleep drifting later and later each day. Individuals with Non-24 have abnormal night sleep patterns, accompanied by difficulty staying awake during the day. Non-24 leads to significant impairment, with chronic effects impacting the social and occupational functioning of these individuals.
In addition to problems sleeping at the desired time, individuals with Non-24 experience excessive daytime sleepiness that often results in daytime napping. TASIMELTION
TASIMELTION
The severity of nighttime sleep complaints and/or daytime sleepiness complaints varies depending on where in the cycle the individual’s body clock is with respect to their social, work, or sleep schedule. The “free running” of the clock results in approximately a 1-4 month repeating cycle, the circadian cycle, where the circadian drive to initiate sleep continually shifts a little each day (about 15 minutes on average) until the cycle repeats itself. Initially, when the circadian cycle becomes desynchronous with the 24 h day-night cycle, individuals with Non-24 have difficulty initiating sleep. As time progresses, the internal circadian rhythms of these individuals becomes 180 degrees out of synchrony with the 24 h day-night cycle, which gradually makes sleeping at night virtually impossible, and leads to extreme sleepiness during daytime hours.
Eventually, the individual’s sleep-wake cycle becomes aligned with the night, and “free-running” individuals are able to sleep well during a conventional or socially acceptable time. However, the alignment between the internal circadian rhythm and the 24-hour day-night cycle is only temporary. In addition to cyclical nighttime sleep and daytime sleepiness problems, this condition can cause deleterious daily shifts in body temperature and hormone secretion, may cause metabolic disruption and is sometimes associated with depressive symptoms and mood disorders.
It is estimated that 50-75% of totally blind people in the United States (approximately 65,000 to 95,000) have Non-24. This condition can also affect sighted people. However, cases are rarely reported in this population, and the true rate of Non-24 in the general population is not known.
The ultimate treatment goal for individuals with Non-24 is to entrain or synchronize their circadian rhythms into an appropriate phase relationship with the 24-hour day so that they will have increased sleepiness during the night and increased wakefulness during the daytime.
INTRODUCTION
Tasimelteon has the chemical name: trans-N-[[2-(2,3-dihydrobenzofuran-4-yl)cycloprop-1yl]methyl]propanamide, has the structure of Formula I:
and is disclosed in U.S. Pat. No. 5,856,529 and in US 20090105333, both of which are incorporated herein by reference as though fully set forth.
Tasimelteon is a white to off-white powder with a melting point of about 78° C. (DSC) and is very soluble or freely soluble in 95% ethanol, methanol, acetonitrile, ethyl acetate, isopropanol, polyethylene glycols (PEG-300 and PEG-400), and only slightly soluble in water. The native pH of a saturated solution of tasimelteon in water is 8.5 and its aqueous solubility is practically unaffected by pH. Tasimelteon has 2-4 times greater affinity for MT2R relative to MT1R. It’s affinity (Ki) for MT1R is 0.3 to 0.4 and for MT2R, 0.1 to 0.2. Tasimelteon is useful in the practice of this invention because it is a melatonin agonist that has been demonstrated, among other activities, to entrain patients suffering from Non-24.
………………………..
SYNTHESIS
(1R-trans)-N-[[2 - (2,3-dihydro-4 benzofuranyl) cyclopropyl] methyl] propanamide PATENT: BRISTOL-MYERS SQUIBB PRIORITY DATE: 1996 HYPNOTIC

PREPARATION OF XV
XXIV D-camphorsulfonic acid IS REACTED WITH THIONYL CHLORIDE TO GIVE
…………XXV (1S, 4R) -7,7-dimethyl-2-oxo-bicyclo [2.2.1] heptane-1-methanesulfonyl chloride
TREATED WITH
XXVI ammonium hydroxide
TO GIVE
XXVII (1S, 4R) -7,7-dimethyl-2-oxo-bicyclo [2.2.1] heptane-1-methanesulfonamide
TREATED WITH AMBERLYST15
….XXVIII (3aS, 6R) -4,5,6,7-tetrahydro-8 ,8-dimethyl-3H-3a ,6-methano-2 ,1-benzisothiazole-2 ,2-dioxide
TREATED WITH LAH, ie double bond is reduced to get
…..XV (3aS, 6R, 7aR)-hexahydro-8 ,8-dimethyl-3H-3a ,6-methano-2 ,1-benzisothiazole-2 ,2-dioxide
Intermediate
I 3-hydroxybenzoic acid methyl ester
II 3-bromo-1-propene
III 3 – (2-propenyloxy) benzoic acid methyl ester
IV 3-hydroxy-2-(2-propenyl) benzoic acid methyl ester
V 2,3-dihydro-4-hydroxy-2-benzofurancarboxylic acid methyl ester
VI benzofuran-4-carboxylic acid methyl ester
VII benzofuran-4-carboxylic acid
VIII 2,3-dihydro-4-benzofurancarboxylic acid
IX 2,3-dihydro-4-benzofuranmethanol
X 2,3-dihydro-4-benzofurancarboxaldehyde
XI Propanedioic acid
XII (E) -3 – (2,3-dihydro-4-benzofuranyl) propenoic acid
XIII thionyl chloride
XIV (E) -3 – (2,3-dihydro-4-benzofuranyl) propenoyl chloride
XV (3aS, 6R, 7aR)-hexahydro-8 ,8-dimethyl-3H-3a ,6-methano-2 ,1-benzisothiazole-2 ,2-dioxide
XVI (3aS,6R,7aR)-1-[(E)-3-(2,3-dihydro-4-benzofuranyl)-1-oxo-2-propenyl]hexahydro-8,8-dimethyl-3H-3a,6-methano-2,1-benzisothiazole-2,2-dioxide
XVII (3aS,6R,7aR)-1-[[(1R,2R)-2-(2,3-dihydro-4-benzofuranyl)cyclopropyl]carbonyl]hexahydro-8,8-dimethyl-3H-3a,6-methano-2,1-benzisothiazole-2,2-dioxide
XVIII [R-(R *, R *)] -2 – (2,3-dihydro-4-benzofuranyl) cyclopropanemethanol
XIX [R-(R *, R *)] -2 – (2,3-dihydro-4-benzofuranyl) cyclopropanecarboxaldehyde
XX hydroxylamine hydrochloride
XXI [R-(R *, R *)] -2 – (2,3-dihydro-4-benzofuranyl) cyclopropanecarbaldehyde oxime
XXII [R-(R *, R *)] -2 – (2,3-dihydro-4-benzofuranyl) cyclopropanemethanamine
XXIII propanoyl chloride
XXIV D-camphorsulfonic acid
XXV (1S, 4R) -7,7-dimethyl-2-oxo-bicyclo [2.2.1] heptane-1-methanesulfonyl chloride
XXVI ammonium hydroxide
XXVII (1S, 4R) -7,7-dimethyl-2-oxo-bicyclo [2.2.1] heptane-1-methanesulfonamide
XXVIII (3aS, 6R) -4,5,6,7-tetrahydro-8 ,8-dimethyl-3H-3a ,6-methano-2 ,1-benzisothiazole-2 ,2-dioxide
Bibliography
- Patents: Benzofuran and dihydrobenzofuran melatonergic agents: US5856529 (1999)
Priority: US19960032689P, 10 Dec. 1996 (Bristol-Myers Squibb Company, U.S.)
- Preparation III (quinazolines): US2004044015 (2004) Priority: EP20000402845, 13 Oct. 2000
- Preparation of VII (aminoalkylindols): Structure-Activity Relationships of Novel Cannabinoid Mimetics Eissenstat et al, J.. Med. Chem. 1995, 38, 3094-3105
- Preparation XXVIII: Towson et al. Organic Syntheses, Coll. Vol. 8, p.104 (1993) Vol. 69, p.158 (1990)
- Preparation XV: Weismiller et al. Organic Syntheses, Coll. Vol. 8, p.110 (1993) Vol. 69, p.154 (1990).
- G. Birznieks et al. Melatonin agonist VEC-162 Improves sleep onset and maintenance in a model of transient insomnia. Sleep 2007, 30, 0773 Abstract.
-. Rajaratnam SM et al, The melatonin agonist VEC-162 Phase time immediately advances the human circadian system, Sleep 2006, 29, 0159 Abstract.
-. AK Singh et al, Evolution of a manufacturing route for a highly potent drug candidate, 229th ACS Natl Meet, March 13-17, 2005, San Diego, Abstract MEDI 576.
- Vachharajani NN et al, Preclinical pharmacokinetics and metabolism of BMS-214778, a novel melatonin receptor agonist, J Pharm Sci. 2003 Apr; 92 (4) :760-72.
. – JW Scott et al, Catalytic Asymmetric Synthesis of a melotonin antagonist; synthesis and process optimization. 223rd ACS Natl Meet, April 7-11, Orlando, 2002, Abstract ORGN 186.
…………………….
SYNTHESIS CONSTRUCTION AS IN PATENT
GENERAL SCHEMES
Reaction Scheme 1
The syntheses of the 4-aryl-propenoic acid derivatives, 2 and 3, are shown in Reaction Scheme 1. The starting aldehydes, 1 , can be prepared by methods well known to those skilled in the art. Condensation of malonic acid with the aldehydes, 1, in solvents such as pyridine with catalysts such as piperidine or pyrrolidine, gives the 4-aryl- propenoic acid, 2. Subsequent conversion of the acid to the acid chloride using reagents such as thionyl chloride, phosphoryl chloride, or the like, followed by reaction with N,0-dimethyl hydroxylamine gives the amide intermediate 3 in good yields. Alternatively, aldehyde 1 can be converted directly to amide 3 using reagents such as diethyl (N-methoxy- N-methyl-carbamoylmethyl)phosphonate with a strong base such as sodium hydride.
Reaction Scheme 2
The conversion of the amide intermediate 3 to the racemic, trans- cyclopropane carboxaldehyde intermediate, 4, is shown in Reaction Scheme 2. Intermediate 3 was allowed to react with cyclopropanating reagents such as trimethylsulfoxonium iodide and sodium hydride in solvents such as DMF, THF, or the like. Subsequent reduction using reagents such as LAH in solvents such as THF, ethyl ether, or the like, gives the racemic, trans-cyclopropane carboxaldehyde intermediates, 4.
Reaction Scheme 3
Racemic cyclopropane intermediate 5 (R = halogen) can be prepared from intermediate 2 as shown in Reaction Scheme 3. Intermediate 2 was converted to the corresponding allylic alcohol by treatment with reducing agents such as sodium borohydride plus iodine in solvents such as THF. Subsequent acylation using reagents such as acetic anhydride in pyridine or acetyl chloride gave the allylic acetate which was allowed to react with cyclopropanating reagents such as sodium chloro-difluoroacetate in diglyme to provide the racemic, trans- cyclopropane acetate intermediates, 5. Reaction Scheme 4
The conversion of the acid 2 to the chiral cyclopropane carboxaldehyde intermediate, (-)-(trans)-4, is shown in Reaction Scheme 4. Intermediate 2 is condensed with (-)-2,10-camphorsultam under standard conditions, and then cyclopropanated in the presence of catalysts such as palladium acetate using diazomethane generated from reagents such as 1-methyl-3-nitro-1-nitrosoguanidine. Subsequent reduction using reagents such as LAH in solvents such as THF, followed by oxidation of the alcohol intermediates using reagents such as DMSO/oxalyl chloride, or PCC, gives the cyclopropane carboxaldehyde intermediate, (-)-(trans)-4, in good yields. The enantiomer, (+)-(trans)-4, can also be obtained employing a similar procedure using (+)-2,10- camphorsultam in place of (-)-2,10-camphorsultam.
When it is desired to prepare compounds of Formula I wherein m = 2, the alcohol intermediate may be activated in the conventional manner such as with mesyl chloride and treated with sodium cyanide followed by reduction of the nitrile group with a reducing agent such as LAH to produce the amine intermediate 6.
Reaction Scheme 5
Reaction Scheme 5 shows the conversion of intermediates 4 and 5 to the amine intermediate, 7, and the subsequent conversion of 6. or 7 to compounds of Formula I. The carboxaldehyde intermediate, 4, is condensed with hydroxylamine and then reduced with reagents such as LAH to give the amine intermediate, 7. The acetate intermediate 5 is hydrolyzed with potassium hydroxide to the alcohol, converted to the mesylate with methane sulfonyl chloride and triethyl amine in CH2CI2and then converted to the azide by treatment with sodium azide in solvents such as DMF. Subsequent reduction of the azide group with a reducing agent such as LAH produced the amine intermediate 7. Further reaction of 6 or 7 with acylating reagents gives compounds of Formula I. Suitable acylating agents include carboxylic acid halides, anhydrides, acyl imidazoles, alkyl isocyanates, alkyl isothiocyanates, and carboxylic acids in the presence of condensing agents, such as carbonyl imidazole, carbodiimides, and the like. Reaction Scheme 6
Reaction Scheme 6 shows the alkylation of secondary amides of Formula I (R2 = H) to give tertiary amides of Formula I (R2 = alkyl). The secondary amide is reacted with a base such as sodium hydride, potassium tert-butoxide, or the like, and then reacted with an alkylating reagent such as alkyl halides, alkyl sulfonate esters, or the like to produce tertiary amides of Formula I.
Reaction Scheme 7
Reaction Scheme 7 shows the halogenation of compounds of Formula I. The carboxamides, i (Q1 = Q2 = H), are reacted with excess amounts of halogenating agents such as iodine, N-bromosuccinimide, or the like to give the dihalo-compounds of Formula I (Q1 = Q2 = halogen). Alternatively, a stoichiometric amount of these halogenating agents can be used to give the monohalo-compounds of Formula I (Q1 = H, Q2 = halogen; or Q1 = halogen, Q2 = H). In both cases, additives such as lead IV tetraacetate can be used to facilitate the reaction. Biological Activity of the Compounds
The compounds of the invention are melatonergic agents. They have been found to bind human melatonergic receptors expressed in a stable cell line with good affinity. Further, the compounds are agonists as determined by their ability, like melatonin, to block the forskolin- stimulated accumulation of cAMP in certain cells. Due to these properties, the compounds and compositions of the invention should be useful as sedatives, chronobiotic agents, anxiolytics, antipsychotics, analgesics, and the like. Specifically, these agents should find use in the treatment of stress, sleep disorders, seasonal depression, appetite regulation, shifts in circadian cycles, melancholia, benign prostatic hyperplasia and related conditions
EXPERIMENTAL PROCEDURES
SEE ORIGINAL PATENT FOR CORECTIONS
Preparation 1
Benzofuran-4-carboxaldehyde
Step 1 : N-Methoxy-N-methyl-benzofuran-4-carboxamide
A mixture of benzofuran-4-carboxylic acid [Eissenstat, et al.. J. Medicinal Chemistry, 38 (16) 3094-3105 (1995)] (2.8 g, 17.4 mmol) and thionyl chloride (25 mL) was heated to reflux for 2 h and then concentrated in vacuo. The solid residue was dissolved in ethyl acetate (50 mL) and a solution of N,O-dimethylhydroxylamine hydrochloride (2.8 g) in saturated NaHC03(60 mL) was added with stirring. After stirring for 1.5 h, the ethyl acetate layer was separated. The aqueous layer was extracted with ethyl acetate. The ethyl acetate extracts were combined, washed with saturated NaHCO3 and concentrated in vacuo to give an oil (3.2 g, 95.4%).
Step 2: Benzofuran-4-carboxaldehyde
A solution of N-methoxy-N-methyl-benzofuran-4-carboxamide (3.2 g, 16.6 mmol) in THF (100 mL) was cooled to -45°C and then LAH (0.7 g, 18.7 mmol) was added. The mixture was stirred for 15 min, allowed to warm to -5°C, and then recooled to -45°C. Saturated KHS04 (25 mL) was added with vigorous stirring, and the mixture was allowed to warm to room temperature. The precipitate was filtered and washed with acetone. The filtrate was concentrated in vacuo to give an oil (2.3 g, 94%). Preparation 2
2,3-Dihydrobenzofuran-4-carboxaldehyde
Step 1 : 2,3-Dihydrobenzofuran-4-carboxylic acid
Benzofuran-4-carboxylic acid (10.0 g, 61 .7 mmol) was hydrogenated (60 psi) in acetic acid (100 mL) over 10% Pd/C (2 g) for 12 hr. The mixture was filtered and the filtrate was diluted with water (500 mL) to give 2,3- dihydrobenzofuran-4-carboxylic acid as a white powder (8.4 g, 83%). A sample was recrystallized from isopropanol to give fine white needles (mp: 185.5-187.5°C).
Step 2: (2,3-Dihydrobenzofuran-4-yl)methanol
A solution of 2,3-dihydrobenzofuran-4-carboxylic acid (10 g, 61 mmol) in THF (100 mL) was stirred as LAH (4.64 g, 122 mmol) was slowly added. The mixture was heated to reflux for 30 min. The mixture was cooled and quenched cautiously with ethyl acetate and then with 1 N HCI (150 mL). The mixture was then made acidic with 12 N HCI until all the inorganic precipitate dissolved. The organic layer was separated, and the inorganic layer was extracted twice with ethyl acetate. The organic layers were combined, washed twice with brine, and then concentrated in vacuo. This oil was Kϋgelrohr distilled to a clear oil that crystallized upon cooling (8.53 g, 87.6%).
Step 3: 2.3-Dihydrobenzofuran-4-carboxaldehyde
DMSO (8.10 mL, 1 14 mmol) was added at -78°C to a stirred solution of oxalyl chloride in CH2CI2 (40 mL of a 2M solution). A solution of (2,3- dihydrobenzofuran-4-yl)methanol (8.53 g, 56.9 mmol) in CH2CI2 (35 mL) was added dropwise, and the solution stirred at -78°C for 30 min. Triethyl amine (33 mL, 228 mmol) was added cautiously to quench the reaction. The resulting suspension was stirred at room temperature for 30 min and diluted with CH2CI2 (100 mL). The organic layer was washed three times with water, and twice with brine, and then concentrated in vacuo to an oil (8.42 g, 100%) that was used without purification.
Preparation 16
(±)-(trans)-2-(2,3-Dihyd robenzofuran-4-yl)cyclopropane- carboxaldehyde
Step 1 : (±Htrans)-N-Methoxy-N-methyl-2-(2.3-dihydrobenzofuran-4- yhcyclopropanecarboxamide
Trimethylsulfoxonium iodide (9.9 g, 45 mmol) was added in small portions to a suspension of sodium hydride (1 .8 g, 45 mmol) in DMF (120 mL). After the foaming had subsided (10 min), a solution of (trans)- N-methoxy-N-methyl-3-(2,3-dihydrobenzofuran-4-yl)propenamide (3.5 g, 15 mmol) in DMF (60 mL) was added dropwise, with the temperature maintained between 35-40°C. The mixture was stirred for 3 h at room temperature. Saturated NH4CI (50 mL) was added dropwise and the mixture was extracted three times with ethyl acetate. The organic extracts were combined, washed with H2O and brine, dried over K2CO3, and concentrated in vacuo to give a white wax (3.7 g, 100%).
Step 2: (±)-(trans)- 2-(2.3-Dihydrobenzofuran-4-yl)cyclopropane- carboxaldehyde
A solution of (±)-(trans)-N-methoxy-N-methyl-2-(2,3-dihydrobenzofuran- 4-yl)cyclopropanecarboxamide (3.7 g, 15 mmol) in THF (10 mL) was added dropwise to a rapidly stirred suspension of LAH (683 mg, 18 mmol) in THF (50 mL) at -45°C, maintaining the temperature below -40°C throughout. The cooling bath was removed, the reaction was allowed to warm to 5°C, and then the reaction was immediately recooled to -45°C. Potassium hydrogen sulfate (3.4 g, 25.5 mmol) in H20 (50 mL) was cautiously added dropwise, the temperature maintained below – 30°C throughout. The cooling bath was removed and the suspension was stirred at room temperature for 30 min. The mixture was filtered through Celite and the filter cake was washed with ether. The combined filtrates were then washed with cold 1 N HCI, 1 N NaOH, and brine. The filtrates were dried over MgSO4, and concentrated in vacuo to give a clear oil (2.6 g, 99%).
Preparation 18
(-)-(trans)-2-(2.3-Dihydrobenzofuran-4-yl)cyclopropane-carboxaldehyde
Step 1 : (-Htrans)-N-[3-(2.3-Dihvdrobenzofuran-4-yl)-propenoyll-2.10- camphorsultam
To a solution of (-)-2,10-camphorsultam (8.15 g, 37.9 mmol) in 50 mL toluene at 0°C was added sodium hydride (1.67 g, 41.7 mmol). After stirring for 0.33 h at 0°C and 0.5 h at 20°C and recooling to 0°C, a solution of 3-(2,3-dihydrobenzofuran-4-yl)-2-propenoyl chloride
(37.9 mmol), prepared in situ from the corresponding acid and thionyl chloride (75 mL), in toluene (50 mL), was added dropwise. After stirring for 18 h at 20°C, the mixture was diluted with ethyl acetate and washed with water, 1 N HCI, and 1 N NaOH. The organic solution was dried and concentrated in vacuo to give 15.8 g of crude product. Recrystallization form ethanol-methanol (600 mL, 1 :1) gave the product (13.5 g, 92%, mp 199.5-200°C).
Step 2: (-)-N-[[(trans)-2-(2,3-Dihydrobenzofuran-4-yl)-cyclopropylj- carbonylj-2, 10-camphorsultam
1 -Methyl-3-nitro-1 -nitrosoguanidine (23.88g 163 mmol) was added in portions to a mixture of 10 N sodium hydroxide (60 mL) and ether (200 mL) at 0°C. The mixture was shaken vigorously for 0.25 h and the ether layer carefully decanted into a solution of (-)-N-[3-(2,3-dihydrobenzofuran-4-yl)-2-propenoyl]-2,10-camphorsultam (9.67 g, 25 mmol) and palladium acetate (35 mg) in methylene chloride (200 mL). After stirring for 18 h, acetic acid (5 mL) was added to the reaction and the mixture stirred for 0.5 h. The mixture was washed with 1 N HCI, 1 N NaOH and brine. The solution was dried, concentrated in vacuo and the residue crystallized twice from ethanol to give the product (6.67 g, 66.5%, mp 157-159°C).
Step 3: (-)-(trans)-2-(2,3-Dihydrobenzofuran-4-yl)cyclopropane- methanol
A solution of (-)-N-[(trans)-2-(2,3-dihydrobenzofuran-4-yl)cyclo-propanecarbonylj-2,10-camphorsultam (4.3 g, 10.7 mmol) in THF (50 mL) was added dropwise to a mixture of LAH (0.81 g, 21.4 mmol) in THF (50 mL) at -45°C. The mixture was stirred for 2 hr while it warmed to 10°C. The mixture was recooled to -40°C and hydrolyzed by the addition of saturated KHS0 (20 mL). The mixture was stirred at room temperature for 30 minutes and filtered. The precipitate was washed twice with acetone. The combined filtrate and acetone washes were concentrated in vacuo. The gummy residue was dissolved in ether, washed with 1 N NaOH and 1 N HCI, and then dried in vacuo to give the product (2.0 g, 98.4%).
Step 4: (-)-(trans)-2-(2.3-Dihydrobenzofuran-4-yl)cyclopropane- carboxaldehyde DMSO (1.6 g, 21 mmol) was added to oxalyl chloride in CH2CI2(7.4 mL of 2 M solution, 14.8 mmole) at -78°C. The (-)-(trans)-2-(2,3-dihydrobenzofuran-4-yl)-cyclopropylmethanol (2.0 g, 10.5 mmol) in CH2CI2(15 mL) was added. The mixture was stirred for 20 min and then triethylamine (4.24 g, 42 mmol) was added. The mixture was warmed to room temperature and stirred for 30 min. The mixture was diluted with CH2CI2 and washed with water, 1 N HCI, and then 1 N NaOH. The organic layer was dried and concentrated iι> vacuo to give the aldehyde product (1.98 g, 100%).
Preparation 24
(-)-(trans)-2-(2.3-Dihydrobenzofuran-4-yl)cyclopropane-methanamine A mixture of (-)-(trans)-2-(2,3-dihydrobenzofuran-4-yl)cyclopropane-carboxaldehyde (1.98 g, 10.5 mmol), hydroxylamine hydrochloride (2.29 g, 33 mmol), and 30% NaOH (3.5 mL, 35 mmol), in 5:1
ethanol/water (50 mL) was heated on a steam bath for 2 h. The solution was concentrated in vacuo. and the residue mixed with water. The mixture was extracted with CH2CI2. The organic extracts were dried and concentrated in vacuo to give a solid which NMR analysis showed to be a mixture of the cis and trans oximes. This material was dissolved in THF (20 mL) and added to solution of alane in THF [prepared from LAH (1.14 g, 30 mmol) and H2S04 (1.47 g, 15 mmol) at 0°Cj. The reaction was stirred for 18 h, and quenched successively with water (1.15 mL), 15% NaOH (1.15 mL), and then water (3.45 mL). The mixture was filtered and the filtrate was concentrated in vacuo. The residue was mixed with ether and washed with water and then 1 N HCI. The acid washes were made basic and extracted with CH2CI . The extracts were dried and concentrated in vacuo to give the amine product (1.4 g, 70.5%). The amine was converted to the fumarate salt in ethanol (mp: 197-198°C).
Anal. Calc’d for C12H15NO • C4H404: C, 62.94; H, 6.27; N, 4.59.
Found: C, 62.87; H, 6.31 ; N, 4.52.
FINAL PRODUCT TASIMELTEON
Example 2
(-)-(trans)-N-[[2-(2,3-Dihydrobenzofuran-4-yl)cycloprop-1-yl]methyl]propanamide
This compound was prepared similar to the above procedure using propionyl chloride and (-)-(trans)-2-(2,3-dihydrobenzofuran-4-yl)- cyclopropanemethanamine to give an oil that solidified upon standing to an off-white solid (61 %, mp: 71-72°C). IR (NaCI Film): 3298, 1645, 1548, 1459, 1235 cm“1.
Mo5 : -17.3°
Anal. Calc’d for C15H19N02: C, 73.44; H, 7.87; N, 5.71 . Found: C, 73.28; H, 7.68; N, 5.58.
Curr Med Chem. 2012;19(21):3532-49. Review.
7 Preclinical pharmacokinetics and metabolism of BMS-214778, a novel melatonin receptor agonist.
Vachharajani NN, Yeleswaram K, Boulton DW.J Pharm Sci. 2003 Apr;92(4):760-72.
 TASIMELTION
TASIMELTION
PATENTS
|
10-15-2010
|
PREDICTION OF SLEEP PARAMETER AND RESPONSE TO SLEEP-INDUCING COMPOUND BASED ON PER3 VNTR GENOTYPE
|
|
|
8-21-2009
|
TREATMENT FOR DEPRESSIVE DISORDERS
|
|
|
5-10-2000
|
Benzopyran derivatives as melatonergic agents
|
|
|
11-10-1999
|
Benzodioxa alkylene ethers as melatonergic agents
|
|
|
6-19-1998
|
BENZODIOXOLE, BENZOFURAN, DIHYDROBENZOFURAN, AND BENZODIOXANE MELATONERGIC AGENTS
|
| WO2007137244A1 * | May 22, 2007 | Nov 29, 2007 | Gunther Birznieks | Melatonin agonist treatment |
| US4880826 | Jun 25, 1987 | Nov 14, 1989 | Nava Zisapel | Melatonin antagonist |
| US4997845 | May 10, 1990 | Mar 5, 1991 | Eli Lilly And Company | β-alkylmelatonins as ovulation inhibitors |
| US5093352 | May 16, 1990 | Mar 3, 1992 | Whitby Research, Inc. | Antidepressant agents |
| US5151446 | Mar 28, 1991 | Sep 29, 1992 | Northwestern University | Substituted 2-amidotetralins as melatonin agonists and antagonists |
| US5225442 | Jan 3, 1992 | Jul 6, 1993 | Adir Et Compagnie | Compounds having a naphthalene structure |
| US5580878 | Jun 7, 1995 | Dec 3, 1996 | Interneuron Pharmaceuticals, Inc. | Substituted tryptamines phenalkylamines and related compounds |
| US5856529 | Dec 9, 1997 | Jan 5, 1999 | Bristol-Myers Squibb Company | Benzofuran and dihydrobenzofuran melatonergic agents |
| US6211225 | Jun 6, 2000 | Apr 3, 2001 | Bristol-Meyers Squibb | Heterocyclic aminopyrrolidine derivatives as melatonergic agents |
| US7754902 | May 18, 2006 | Jul 13, 2010 | Vanda Pharmaceuticals, Inc. | Ruthenium(II) catalysts for use in stereoselective cyclopropanations |
| US20010047016 | Apr 12, 2001 | Nov 29, 2001 | Gregory Oxenkrug | Method for treating depression |
| US20050164987 | Dec 22, 2004 | Jul 28, 2005 | Barberich Timothy J. | Melatonin combination therapy for improving sleep quality |
| US20090105333 | May 22, 2007 | Apr 23, 2009 | Gunther Birznieks | Melatonin agonist treatment |
extra info
|
 |

dedicated to lionel my son



my daughter Aishal

THEY KEEP ME GOING
Selinexor (KPT-330)
1393477-72-9
WO2011109799A1
synthesis at http://www.allfordrugs.com/2014/06/10/karyopharm-announces-initiation-of-phase-2-study-of-selinexor-kpt-330/
Karyopharm Announces Initiation of Phase 2 Study of Selinexor (KPT-330) in Patients with …
MarketWatch
“These patients were treated in our Phase 1 clinical trial of Selinexor in … Additional Phase 1 and Phase 2 studies are ongoing or currently planned and … the discovery and development of novel first-in-class drugs directed against …
synthesis
THANKS AND REGARD’S
DR ANTHONY MELVIN CRASTO Ph.D
amcrasto@gmail.com
VIETNAM
http://me.zing.vn/u/amcrasto
ICELAND
http://amcrasto.bland.is/
RUSSIA
http://www.100zakladok.ru/amcrasto/
http://bobrdobr.ru/people/amcrasto/


US Orphan Drug Market Outlook 2018
Academia.edu
US Orphan Drug Pipeline Insight by Phase & Indication 5.1 Research 5.2 Preclinical 5.3 Phase I 5.4 Phase I/II 5.5 Phase II 5.6 Phase II/III 5.7 Phase III …

http://www.academia.edu/7453102/US_Orphan_Drug_Market_Outlook_2018 …………… download at this site
![]()
Market Overview
In the largest market for orphan drugs, USA, there was a shortage of adequate therapies for treating many rare diseases. These therapies were not developed as companies did not expect these drugs to be highly profitable. Hence there was a lack of interest and thus investment on the part of pharma companies in the USA. Therefore, the FDA introduced incentives for developing such drugs. This step taken by the FDA was successful in creating a thriving market for orphan drugs. It was in the USA first that a special law exclusively for governing orphan drugs was framed in the form of the Orphan Drug Act of 1983. This led to an increase in the popularity of orphan drugs. The FDA also has been continuously increasing its efforts to support this market by providing significant financial and non-financial incentives to the pharmaceutical companies to attract them. This has been one of the major drivers of growth for the US orphan drugs market.
Figure 3-1: US Orphan Drug Market (US$ Billion), 2012-2018
2012201320142015201620172018
Source: KuicK Research
see my profile
http://ictmumbai.academia.edu/AnthonyMelvinCrastoPhD


ATALUREN
PTC 124
3-[5-(2-Fluorophenyl)-1,2,4-oxadiazol-3-yl]benzoic acid
| MF C15H9FN2O3 | ||
| Molecular Weight | 284.24 | |
| CAS Registry Number | 775304-57-9 |
PTC Therapeutics Initiates Confirmatory Phase 3 Clinical Trial of Translarna™ (ataluren) in Patients with Nonsense Mutation Cystic Fibrosis (nmCF) – MarketWatch

Ataluren, formerly known as PTC124, is a small-molecular agent designed by PTC Therapeutics and sold under the trade nameTranslarna. It makes ribosomes less sensitive to premature stop codons (referred to as “read-through”). This may be beneficial in diseases such as Duchenne muscular dystrophy where the mRNA contains a mutation causing premature stop codons or nonsense codons. There is ongoing debate over whether Ataluren is truly a functional drug (inducing codon read-through), or if it is nonfunctional, and the result was a false-positive hit from a biochemical screen based on luciferase.[1]
Ataluren has been tested on healthy humans and humans carrying genetic disorders caused by nonsense mutations,[2][3] such as some people with cystic fibrosis and Duchenne muscular dystrophy. In 2010, PTC Therapeutics released preliminary results of its phase 2b clinical trial for Duchenne muscular dystrophy, with participants not showing a significant improvement in the six minute walk distance after the 48 weeks of the trial.[4] This failure resulted in the termination of a $100 million deal with Genzyme to pursue the drug. However, other phase 2 clinical trials were successful for cystic fibrosis in Israel, France and Belgium.[5] Multicountry phase 3 clinical trials are currently in progress for cystic fibrosis in Europe and the USA.[6]
In cystic fibrosis, early studies of ataluren show that it improves nasal potential difference.[7]
Ataluren appears to be most effective for the stop codon ‘UGA’.[2]
On 23 May 2014 ataluren received a positive opinion from the Committee for Medicinal Products for Human Use (CHMP) of the European Medicines Agency (EMA).[8]
It is not that ataluren is a complex molecule. To judge from one of the patents, synthesis is straightforward starting from 2-cyanobenoic acid and 2-fluorobenzoyl chloride, both commercially available. The synthetic steps are methylation of 2-cyanobenoic acid (iodomethane), nitrile hydrolysis with hydroxylamine, esterification with the fluoro acid chloride using DIPEA, high-temperature dehydration to the oxadiazole and finally ester hydrolysis (NaOH).


other sources
Ataluren Molecule
Orphan drug under investigation for treatment of genetic conditions where nonsense mutations result in premature termination of polypeptides. This drug, which is convenient to deliver orally, appears to allow ribosomal transcription ofRNA to continue past premature termination codon mutations with correct reading of the full normal transcript which then terminates at the proper stop codon. Problematically it has been postulated that assay artifact may have complicated evaluation of its efficacy which appears to be less than gentamicin.[1] Faults of this class in the transcription process are involved in several inherited diseases.
Some forms of cystic fibrosis and Duchenne muscular dystrophy are being targeted in the development stage of the drug.[2] Phase I and II trials are promising for cystic fibrosis.[3][4] In a mouse model of Duchenne muscular dystrophy, restoration of muscle function occurred.[5]
A potential issue is that there may be parts of the human genome whose optimal gene function through evolution has resulted from relatively recent in evolutionary terms insertion of a premature termination codon and so functional suboptimal transcripts of other proteins or functional RNAs might result.
old cut paste
A large-scale, multinational, phase 3 trial of the experimental drug ataluren has opened its first trial site, in Cincinnati, Ohio.
The trial is recruiting boys with Duchenne muscular dystrophy (DMD) or Becker muscular dystrophy (BMD) caused by anonsense mutation — also known as a premature stop codon — in the dystrophin gene. This type of mutation causes cells to stop synthesizing a protein before the process is complete, resulting in a short, nonfunctional protein. Nonsense mutations are believed to cause DMD or BMD in approximately 10 to 15 percent of boys with these disorders.
Ataluren — sometimes referred to as a stop codon read-through drug — has the potential to overcome the effects of a nonsense mutation and allow functional dystrophin — the muscle protein that’s missing in Duchenne MD and deficient in Becker MD — to be produced.
The orally delivered drug is being developed by PTC Therapeutics, a South Plainfield, N.J., biotechnology company, to whichMDA gave a $1.5 million grant in 2005.
PTC124 has been developed by PTC Therapeutics.
Dantrolene sodium
1-[[[5-(4-nitrophenyl)-2-furanyl]methylene]amino]-2,4-imidazolidinedione
VIEW THIS POST AT BELOW LINK UNTIL FORMATTING IS FIXED
http://www.allfordrugs.com/2014/07/24/fda-approves-ryanodex-for
-the-treatment-of-malignant-hyperthermia/
FDA Approves Ryanodex for the Treatment of Malignant Hyperthermia
WOODCLIFF LAKE, N.J.(BUSINESS WIRE) July 23, 2014 —
Eagle Pharmaceuticals, Inc. (“Eagle” or “the Company”)
(Nasdaq:EGRX) today announced that the U. S. Food and Drug Administration (FDA)
has approved Ryanodex (dantrolene sodium) for injectable
suspension indicated for
the treatment of malignant hyperthermia (MH), along
with the appropriate supportive measures.
MH is an inherited and potentially fatal disorder triggered
by certain anesthesia agents
in genetically susceptible individuals. FDA had designated
Ryanodex as an Orphan Drug in
August 2013. Eagle has been informed by the FDA that it will learn over the next four to
six weeks if it has been granted the seven year Orphan Drug market exclusivity.
read at
http://www.drugs.com/newdrugs/fda-approves-ryanodex-malignant-
hyperthermia-4058.html?utm_source=ddc&utm_medium=email&utm_
news+summary+-+July+23%2C+2014
READ MORE AT
PATENTS, CAS NO ETC
http://www.allfordrugs.com/2014/07/24/fda-approves-ryanodex-
Migalastat hydrochloride
CAS Number: 75172-81-5 hydrochloride
CAS BASE….108147-54-2
ABS ROT = (+)
|
Conc: 1 g/100mL; Solv: water ; 589.3 nm; Temp: 24 °C |
IN Van den Nieuwendijk, Adrianus M. C. H.; Organic Letters 2010, 12(17), 3957-3959
3,4,5-Piperidinetriol,2-(hydroxymethyl)-, hydrochloride (1:1), (2R,3S,4R,5S)-
Molecular Structure:
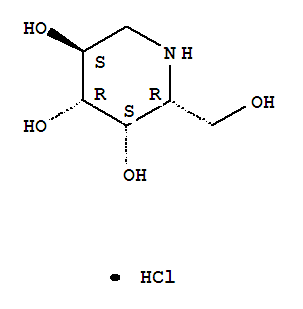
Formula: C6H14ClNO4
Molecular Weight:199.63
Synonyms: 3,4,5-Piperidinetriol,2-(hydroxymethyl)-, hydrochloride, (2R,3S,4R,5S)- (9CI);
3,4,5-Piperidinetriol,2-(hydroxymethyl)-, hydrochloride, [2R-(2a,3a,4a,5b)]-;
Migalastat hydrochloride;Galactostatin hydrochloride;
(2S,3R,4S,5S)-2-(hydroxymethyl)piperidine-3,4,5-triol hydrochloride;
Melting Point:160 °C-162…….http://www.google.com/patents/DE3906463A1?cl=de
Boiling Point:382.7 °C at 760 mmHg
Flash Point:185.2 °C
Amicus Therapeutics, Inc. innovator
Aug 2014
Amicus Therapeutics was on the ropes in late 2012 when its pill for a rare condition called Fabry Disease108147-54-2 failed a late-stage trial. It had already put seven years of work into the drug, and the setback added even more development time and uncertainty to the mix. But the Cranbury, NJ-based company kept plugging away, and now it looks like all the effort could lead to its first approved drug.
Amicus (NASDAQ: FOLD) is reporting today that the Fabry drug, migalastat, succeeded in the second of two late-stage trials. It hit two main goals that essentially measured its ability to slow the decline of Fabry patients’ kidney function comparably to enzyme-replacement therapy (ERT)—the standard of care for the often-fatal disorder.
Amicus believes the results, along with those from an earlier Phase 3 trial comparing migalastat to a placebo, are good enough to ask regulators in the U.S. and Europe for market approval.
“These are the good days to be a CEO,” says Amicus CEO John Crowley (pictured above). “It’s great when a plan comes together and data cooperates.”
Crowley says Amicus will seek approval of migalastat first in Europe and is already in talks with regulators there. In the next few months, Amicus will begin talking with the FDA about a path for approval in the U.S. as well.
End feb 2013
About Amicus Therapeutics
Amicus Therapeutics is a biopharmaceutical company at the forefront of therapies for rare and orphan diseases. The Company is developing orally-administered, small molecule drugs called pharmacological chaperones, a novel, first-in-class approach to treating a broad range of human genetic diseases. Amicus’ late-stage programs for lysosomal storage disorders include migalastat HCl monotherapy in Phase 3 for Fabry disease; migalastat HCl co-administered with enzyme replacement therapy (ERT) in Phase 2 for Fabry disease; and AT2220 co-administered with ERT in Phase 2 for Pompe disease.
About Migalastat HCl
Amicus in collaboration with GlaxoSmithKline (GSK) is developing the investigational pharmacological chaperone migalastat HCl for the treatment of Fabry disease. Amicus has commercial rights to all Fabry products in the United States and GSK has commercial rights to all of these products in the rest of world.
As a monotherapy, migalastat HCl is designed to bind to and stabilize, or “chaperone” a patient’s own alpha-galactosidase A (alpha-Gal A) enzyme in patients with genetic mutations that are amenable to this chaperone in a cell-based assay. Migalastat HCl monotherapy is in Phase 3 development (Study 011 and Study 012) for Fabry patients with genetic mutations that are amenable to this chaperone monotherapy in a cell-based assay. Study 011 is a placebo-controlled study intended primarily to support U.S. registration, and Study 012 compares migalastat HCl to ERT to primarily support global registration.
For patients currently receiving ERT for Fabry disease, migalastat HCl in combination with ERT may improve ERT outcomes by keeping the infused alpha-Gal A enzyme in its properly folded and active form thereby allowing more active enzyme to reach tissues.2Migalastat HCl co-administered with ERT is in Phase 2 (Study 013) and migalastat HCl co-formulated with JCR Pharmaceutical Co. Ltd’s proprietary investigational ERT (JR-051, recombinant human alpha-Gal A enzyme) is in preclinical development.
About Fabry Disease
Fabry disease is an inherited lysosomal storage disorder caused by deficiency of an enzyme called alpha-galactosidase A (alpha-Gal A). The role of alpha-Gal A within the body is to break down specific lipids in lysosomes, including globotriaosylceramide (GL-3, also known as Gb3). Lipids that can be degraded by the action of α-Gal are called “substrates” of the enzyme. Reduced or absent levels of alpha-Gal A activity leads to the accumulation of GL-3 in the affected tissues, including the kidneys, heart, central nervous system, and skin. This accumulation of GL-3 is believed to cause the various symptoms of Fabry disease, including pain, kidney failure, and increased risk of heart attack and stroke.
It is currently estimated that Fabry disease affects approximately 5,000 to 10,000 people worldwide. However, several literature reports suggest that Fabry disease may be significantly under diagnosed, and the prevalence of the disease may be much higher.
2. Benjamin, et al., Molecular Therapy: April 2012, Vol. 20, No. 4, pp. 717–726.
http://clinicaltrials.gov/show/NCT01458119
http://www.docstoc.com/docs/129812511/migalastat-hcl
Migalastat hydrochloride is a pharmacological chaperone in phase III development at Amicus Pharmaceuticals for the oral treatment of Fabry’s disease. Fabry’s disease occurs as the result of an inherited genetic mutation that results in the production of a misfolded alpha galactosidase A (alpha-GAL) enzyme, which is responsible for breaking down globotriaosylceramide (GL-3) in the lysosome. Migalastat acts by selectively binding to the misfolded alpha-GAL, increasing its stability and promoting proper folding, processing and trafficking of the enzyme from the endoplasmic reticulum to the lysosome.
In February 2004, migalastat hydrochloride was granted orphan drug designation by the FDA for the treatment of Fabry’s disease.
The EMEA assigned orphan drug designation for the compound in 2006 for the treatment of the same indication. In 2007, the compound was licensed to Shire Pharmaceuticals by Amicus Therapeutics worldwide, with the exception of the U.S., for the treatment of Fabry’s disease.
In 2009, this license agreement was terminated. In 2010, the compound was licensed by Amicus Therapeutics to GlaxoSmithKline on a worldwide basis to develop, manufacture and commercialize migalastat hydrochloride as a treatment for Fabry’s disease, but the license agreement terminated in 2013.
| Chemical Name: | DEOXYGALACTONOJIRIMYCIN, HYDROCHLORIDE |
| Synonyms: | DGJ;Amigal;Unii-cly7m0xd20;GALACTOSTATIN HCL;DGJ, HYDROCHLORIDE;Migalastat hydrochloride;Galactostatin hydrochloride;DEOXYGALACTONOJIRIMYCIN HCL;1-DEOXYGALACTONOJIRIMYCIN HCL;1,5-dideoxy-1,5-imino-d-galactitol |
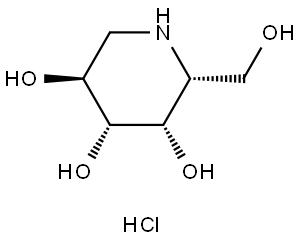
………………………..
http://www.google.co.in/patents/WO1999062517A1?cl=en
Example 1
A series of plant alkaloids (Scheme 1, ref. 9) were used for both in vitro inhibition and intracellular enhancement studies of α-Gal A activity. The results of inhibition experiments are shown in Fig. 1 A.
f^
Among the tested compounds, 1-deoxy-galactonojirimycin (DGJ, 5) known as a powerful competitive inhibitor for α-Gal A, showed the highest inhibitory activity with IC50 at 4.7 nM. α-3,4-Di-epi-homonojirimycin (3) was an effective inhibitor with IC50 at 2.9 μM. Other compounds showed moderate inhibitory activity with IC50 ranging from 0.25 mM (6) to 2.6 mM (2). Surprisingly, these compounds also effectively enhanced α-Gal A activity in COS-1 cells transfected with a mutant α-Gal A gene (R301Q), identified from an atypical variant form of Fabry disease with a residual α- Gal A activity at 4% of normal. By culturing the transfected COS-1 cells with these compounds at concentrations cat 3 – 10-fold of IC50 of the inhibitors, α-Gal A activity was enhanced 1.5 – 4-fold (Fig. 1C). The effectiveness of intracellular enhancement paralleled with in vitro inhibitory activity while the compounds were added to the culture medium at lOμM
concentration (Fig. IB).
………………………
WO 2008045015
or http://www.google.com/patents/EP2027137A1?cl=en, http://www.google.com/patents/US7973157?cl=en
This invention relates to a process for purification of imino or amino sugars, such as D-1-deoxygalactonojirimycin hydrochloride (DGJ’HCl). This process can be used to produce multi-kilogram amounts of these nitrogen-containing sugars.
Sugars are useful in pharmacology since, in multiple biological processes, they have been found to play a major role in the selective inhibition of various enzymatic functions. One important type of sugars is the glycosidase inhibitors, which are useful in treatment of metabolic disorders. Galactosidases catalyze the hydrolysis of glycosidic linkages and are important in the metabolism of complex carbohydrates. Galactosidase inhibitors, such as D-I- deoxygalactonojirimycin (DGJ), can be used in the treatment of many diseases and conditions, including diabetes (e.g., U.S. Pat. 4,634,765), cancer (e.g., U.S. Pat. 5,250,545), herpes (e.g. , U.S. Pat. 4,957,926), HIV and Fabry Disease (Fan et al, Nat. Med. 1999 5:1, 112-5).
Commonly, sugars are purified through chromatographic separation. This can be done quickly and efficiently for laboratory scale synthesis, however, column chromatography and similar separation techniques become less useful as larger amounts of sugar are purified. The size of the column, amount of solvents and stationary phase (e.g. silica gel) required and time needed for separation each increase with the amount of product purified, making purification from multi-kilogram scale synthesis unrealistic using column chromatography.
Another common purification technique for sugars uses an ion- exchange resin. This technique can be tedious, requiring a tedious pre-treatment of the ion exchange resin. The available ion exchange resins are also not necessarily able to separate the sugars from salts (e.g., NaCl). Acidic resins tend to remove both metal ions found in the crude product and amino- or imino-sugars from the solution and are therefore not useful. Finding a resin that can selectively remove the metal cations and leave amino- or imino-sugars in solution is not trivial. In addition, after purification of a sugar using an ion exchange resin, an additional step of concentrating the diluted aqueous solution is required. This step can cause decomposition of the sugar, which produces contaminants, and reduces the yield.
U.S. Pats. 6,740,780, 6,683,185, 6,653,482, 6,653,480, 6,649,766, 6,605,724, 6,590,121, and 6,462,197 describe a process for the preparation of imino- sugars. These compounds are generally prepared from hydroxyl-protected oxime intermediates by formation of a lactam that is reduced to the hexitol. However, this process has disadvantages for the production on a multi-kg scale with regard to safety, upscaling, handling, and synthesis complexity. For example, several of the disclosed syntheses use flash chromatography for purification or ion-exchange resin treatment, a procedure that is not practicable on larger scale. One particularly useful imino sugar is DGJ. There are several DGJ preparations disclosed in publications, most of which are not suitable for an industrial laboratory on a preparative scale (e.g., >100 g). One such synthesis include a synthesis from D-galactose (Santoyo-Gonzalez, et al, Synlett 1999 593-595; Synthesis 1998 1787-1792), in which the use of chromatography is taught for the purification of the DGJ as well as for the purification of DGJ intermediates. The use of ion exchange resins for the purification of DGJ is also disclosed, but there is no indication of which, if any, resin would be a viable for the purification of DGJ on a preparative scale. The largest scale of DGJ prepared published is 13 g (see Fred-Robert Heiker, Alfred Matthias Schueller, Carbohydrate Research, 1986, 119-129). In this publication, DGJ was isolated by stirring with ion-exchange resin Lewatit MP 400 (OH“) and crystallized with ethanol. However, this process cannot be readily scaled to multi- kilogram quantities.
Similarly, other industrial and pharmaceutically useful sugars are commonly purified using chromatography and ion exchange resins that cannot easily be scaled up to the purification of multi-kilogram quantities.
Therefore, there is a need for a process for purifying nitrogen- containing sugars, preferably hexose amino- or imino-sugars that is simple and cost effective for large-scale synthesis
FIG. 1. HPLC of purified DGJ after crystallization. The DGJ is over 99.5% pure.

FIG. 2A. 1H NMR of DGJ (post HCl extraction and crystallization), from 0 – 15 ppm in DMSO.

FIG. 2B. 1H NMR of DGJ (post HCl extraction and crystallization), from 0 – 5 ppm, in DMSO.

FIG. 3 A. 1H NMR of purified DGJ (after recrystallization), from 0 – 15 ppm, in D2O. Note OH moiety has exchanged with OD.

FIG. 3B. 1H NMR of purified DGJ (after recrystallization), from 0 -
4 ppm, in D2O. Note OH moiety has exchanged with OD.

FIG. 4. 13C NMR of purified DGJ, (after recrystallization), 45 – 76 ppm.

One amino-sugar of particular interest for purification by the method of the current invention is DGJ. DGJ, or D-l-deoxygalactonojirimycin, also described as (2R,3S,4R,5S)-2-hydroxymethyl-3,4,5-trihydroxypiperidine and 1- deoxy-galactostatin, is a noj irimycin (5-amino-5-deoxy-D-galactopyranose) derivative of the form:
Example 1: Preparation and Purification of DGJ
A protected crystalline galactofuranoside obtained from the technique described by Santoyo-Gonzalez. 5-azido-5-deoxy-l,2,3,6-tetrapivaloyl-α-D- galactofuranoside (1250 g), was hydrogenated for 1-2 days using methanol (10 L) with palladium on carbon (10%, wet, 44 g) at 50 psi of H2. Sodium methoxide (25% in methanol, 1.25 L) was added and hydrogenation was continued for 1-2 days at 100 psi ofH2. Catalyst was removed by filtration and the reaction was acidified with methanolic hydrogen chloride solution (20%, 1.9 L) and concentrated to give crude mixture of DGJ • HCl and sodium chloride as a solid. The purity of the DGJ was about 70% (w/w assay), with the remaining 30% being mostly sodium chloride.
The solid was washed with tetrahydrofuran (2 x 0.5 L) and ether (I x 0.5 L), and then combined with concentrated hydrochloric acid (3 L). DGJ went into solution, leaving NaCl undissolved. The obtained suspension was filtered to remove sodium chloride; the solid sodium chloride was washed with additional portion of hydrochloric acid (2 x 0.3 L). All hydrochloric acid solution were combined and slowly poured into stirred solution of tetrahydrofuran (60 L) and ether (11.3 L). The precipitate formed while the stirring was continued for 2 hours. The solid crude DGJ* HCl, was filtered and washed with tetrahydrofuran (0.5 L) and ether (2 x 0.5 L). An NMR spectrum is shown in FIGS. 2A-2B.
The solid was dried and recrystallized from water (1.2 mL /g) and ethanol (10 ml/1 ml of water). This recrystallization step may be repeated. This procedure gave white crystalline DGJ* HCl, and was usually obtained in about 70- 75% yield (320 – 345 g). The product of the purification, DGJ-HCl is a white crystalline solid, HPLC >98% (w/w assay) as shown in FIG. 1. FIGS. 3A-3D and FIG. 4 show the NMR spectra of purified DGJ, showing the six sugar carbons.
Example 2: Purification of 1-deoxymannojirimycin 1 -deoxymannojirimycin is made by the method described by Mariano
(J. Org. Chem., 1998, 841-859, see pg. 859, herein incorporated by reference). However, instead of purification by ion-exchange resin as described by Mariano, the 1-deoxymannojirimycin is mixed with concentrated HCl. The suspension is then filtered to remove the salt and the 1-deoxymannojirimycin hydrochloride is precipitated crystallized using solvents known for recrystallization of 1- deoxymannojirimycin (THF for crystallization and then ethanol/water.
Example 3: Purification of (+)-l-deoxynojirimycin
(+)-l-deoxynojirimycin is made by the method Kibayashi et al. (J. Org. Chem., 1987, 3337-3342, see pg. 334I5 herein incorporated by reference). It is synthesized from a piperidine compound (#14) in HCl/MeOH. The reported yield of 90% indicates that the reaction is essentially clean and does not contain other sugar side products. Therefore, the column chromatography used by Kibayashi is for the isolation of the product from non-sugar related impurities. Therefore, instead of purification by silica gel chromatography, the (+)-l-deoxynojirimycin is mixed with concentrated HCl. The suspension is then filtered to remove the salt and the nojirimycin is crystallized using solvents known for recrystallization of nojirimycin.
Example 4: Purification of Nojirimycin
Nojirimycin is made by the method described by Kibayashi et al. (J.
Org. Chem., 1987, 3337-3342, see pg. 3342). However, after evaporating of the mixture at reduced pressure, instead of purification by silica gel chromatography with ammonia-methanol-chloroform as described by Kibayashi, the nojirimycin is mixed with concentrated HCl. The suspension is then filtered to remove the impurities not dissolved in HCl and the nojirimycin is crystallized using solvents known for recrystallization of nojirimycin.
……………………….
Synthesis of (+)-1-deoxygalactonojirimycin and a related indolizidine
Tetrahedron Lett 1995, 36(5): 653
Original Research Article
Amido-alcohol 1 is transformed via aminal 2 into 1-deoxygalactonojirimycin (3) and indolizidine 4.
Amido-alcohol 1 is transformed via aminal 2 into 1-deoxygalactonojirimycin (3) and the structurally related indolizidine 4.
………………………
Synthesis of D-galacto-1-deoxynojirimycin (1,5-dideoxy-1,5-imino-D-galactitol) starting from 1-deoxynojirimycin
Carbohydr Res 1990, 203(2): 314
………………………………..
Synthesis of (+)-1,5-dideoxy-1,5-imino-D-galactitol, a potent alpha-D-galactosidase inhibitor
Carbohydr Res 1987, 167: 305
……………………………..
SEE
Monosaccharides containing nitrogen in the ring, XXXVII. Synthesis of 1,5-didexy-1,5-imino-D-galactitol
Chem Ber 1980, 113(8): 2601
…………………………
http://pubs.acs.org/doi/abs/10.1021/ol101556k
|
Conc: 1 g/100mL; Solv: water ; 589.3 nm; Temp: 24 °C |
IN
van den Nieuwendijk, Adrianus M. C. H.; Organic Letters 2010, 12(17), 3957-3959
The chemoenzymatic synthesis of three 1-deoxynojirimycin-type iminosugars is reported. Key steps in the synthetic scheme include a Dibal reduction−transimination−sodium borohydride reduction cascade of reactions on an enantiomerically pure cyanohydrin, itself prepared employing almond hydroxynitrile lyase (paHNL) as the common precursor. Ensuing ring-closing metathesis and Upjohn dihydroxylation afford the target compounds.
http://pubs.acs.org/doi/suppl/10.1021/ol101556k/suppl_file/ol101556k_si_002.pdf
COMPD 18
D-galacto-1-deoxynojirimicin.HCl (18).
D-N-Boc-6-OBn-galacto-1-deoxynojirimicin (159 mg, 0.450 mmol) was dissolved in a mixture of MeOH
(10 mL) and 6 M HCl (2 mL). The flask was purged with argon, Pd/C-10% (20 mg) was added and a balloon
with hydrogen gas was placed on top of the reaction. The mixture was stirred overnight at room temperature.
Pd/C was removed by filtration and the filtrate evaporated to yield the crude product (90 mg, 100%) as a
white foam that needed no further purification.
[α]24D = + 53.0 (c = 1, H2O);
[lit4a [α]24D = +44.6 (c = 0.9, H2O); lit4b [α]20D = +46.1 (c = 0.9, H2O)].
HRMS calculated for [C6H13NO4 + H]+164.09173; Found 164.09160.
1H NMR (400 MHz, D2O) δ 4.20 (dd, J = 2.7, 1.1 Hz, 1H), 4.11 (ddd, J = 11.4, 9.7, 5.4 Hz, 1H), 3.88 (ddd,
J = 20.9, 12.2, 6.8 Hz, 2H), 3.68 (dd, J = 9.7, 3.0 Hz, 1H), 3.55 (dd, J = 12.5, 5.4 Hz, 1H), 3.46 (ddd, J = 8.6,
4.8, 1.0 Hz, 1H), 2.97 – 2.86 (t, J = 12.0 Hz, 1H). [lit4c supporting information contains 1
H NMR-spectrumof an authentic sample].
13C NMR (101 MHz, D2O) δ 73.01, 66.97, 64.69, 60.16, 59.15, 46.15
4a) Ruiz, M.; Ruanova, T. M.; Blanco, O.; Núñez, F.; Pato, C.; Ojea, V. J. Org. Chem. 2008, 73, 2240
– 2255.
4b) Paulsen, H.; Hayauchi, Y.; Sinnwell, V. Chem. Ber. 1980, 113, 2601 – 2608. c)
McDonnell, C.; Cronin, L.; O’Brien, J. L.; Murphy, P. V. J. Org. Chem. 2004, 69, 3565 – 3568.
……………………………………….
(- ) FORM………… BE CAREFUL
Short and straightforward synthesis of (-)-1-deoxygalactonojirimycin
Org Lett 2010, 12(6): 1145
http://pubs.acs.org/doi/abs/10.1021/ol100037c
The mildness and low basicity of vinylzinc species functioning as a nucleophile in addition to α-chiral aldehydes is characterized by lack of epimerization of the vulnerable stereogenic center. This is demonstrated by a highly diastereoselective synthesis of 1-deoxygalactonojirimycin in eight steps from commercial starting materials with overall yield of 35%.
Figure 1. Structures of nojirimycin (1) and DGJ (2).
SEE SUPP INFO
http://pubs.acs.org/doi/suppl/10.1021/ol100037c/suppl_file/ol100037c_si_001.pdf
(-)-1-deoxygalactojirimycin hydrochloride as transparent colorless needles.
[α]D -51.4 (D2O, c 1.0)
1H-NMR (D2O) δ ppm 4.09 (dd, 1H, J 2.9 Hz, 1.3 Hz), 4.00 (ddd, 1H, J = 11.3 Hz, 9.7 Hz, 5.3 Hz),
3.80 (dd, 1H, J = 12,1 Hz, 8.8 Hz), 3.73 (dd, 1H, J = 12.1 Hz, 8.8 Hz), 3.56 (dd, 1H, J = 9.7 Hz, 2.9
Hz), 3.44 (dd, 1H, J = 12.4 Hz, 5.3 Hz), 3.34 (ddd, 1H, J = 8.7 Hz, 4.8 Hz, 1.0 Hz), 2.8 (app. t, 1H,
J = 12.0 Hz)
13C-NMR (D2O, MeOH iSTD) δ 73.6, 67.5, 65.3, 60.7, 59.7, 46.7
HRMS Measured 164.0923 (M + H – Cl) Calculated 164.0923 (C6H13NO4 + H – Cl)
…………………………………………………..
Concise and highly stereocontrolled synthesis of 1-deoxygalactonojirimycin and its congeners using dioxanylpiperidene, a promising chiral building block
Org Lett 2003, 5(14): 2527
http://pubs.acs.org/doi/abs/10.1021/ol034886y
A concise and stereoselective synthesis of the chiral building block, dioxanylpiperidene 4 as a precursor for deoxyazasugars, starting from the Garner aldehyde 5 using catalytic ring-closing metathesis (RCM) for the construction of the piperidine ring is described. The asymmetric synthesis of 1-deoxygalactonojirimycin and its congeners 1−3 was carried out via the use of 4in a highly stereocontrolled mode.
mp 135-135.5 °C [lit.3mp 137-139 °C];
[α]D25 +27.8° (c 0.67, H2O)
[lit.3[α]D23 +28° (c 0.5, H2O)];
1H NMR (300 MHz, D2O) δ 2.59–2.65 (m, 1H), 2.81–2.87 (m, 1H),
3.02–3.08 (m, 1H), 3.46–3.48 (m, 2H), 3.59–3.66 (m, 3H); 13C NMR (75 MHz, D2O) δ 44.7, 57.1,
58.4, 70.9, 71.4, 73.3 [lit4 13C NMR (125 MHz, D2O) δ 44.5, 56.8, 58.3, 70.1, 70.7, 72.3];
HRMScalcd for C6H13NO4 (M+) 163.0855, Found 163.0843. Anal. calcd for C6H13NO4: C, 44.16; N,
8.58; H, 8.03. Found: C, 44.31; N, 8.55; H, 7.71.
3. Schaller, C.; Vogel, P.; Jager, V. Carbohydrate Res. 1998, 314, 25-35.
4. Lee, B. W.; Jeong, Ill-Y.; Yang, M. S.; Choi, S. U.; Park, K. H. Synthesis 2000, 1305-1309.
…………………………………………..
Applications and limitations of the I2-mediated carbamate annulation for the synthesis of piperidines: Five- versus six-membered ring formation
J Org Chem 2013, 78(19): 9791
http://pubs.acs.org/doi/abs/10.1021/jo401512h
A protecting-group-free synthetic strategy for the synthesis of piperidines has been explored. Key in the synthesis is an I2-mediated carbamate annulation, which allows for the cyclization of hydroxy-substituted alkenylamines into piperidines, pyrrolidines, and furans. In this work, four chiral scaffolds were compared and contrasted, and it was observed that with both d-galactose and 2-deoxy-d-galactose as starting materials, the transformations into the piperidines 1-deoxygalactonorjirimycin (DGJ) and 4-epi-fagomine, respectively, could be achieved in few steps and good overall yields. When d-glucose was used as a starting material, only the furan product was formed, whereas the use of 2-deoxy-d-glucose resulted in reduced chemo- and stereoselectivity and the formation of four products. A mechanistic explanation for the formation of each annulation product could be provided, which has improved our understanding of the scope and limitations of the carbamate annulation for piperidine synthesis.
……………………………………………
Ruiz, Maria; Journal of Organic Chemistry 2008, 73(6), 2240-2255
http://pubs.acs.org/doi/abs/10.1021/jo702601z
ROT +44.6 ° Conc: 0.9 g/100mL; Solv: water ; 589.3 nm; Temp: 24 °C
A general strategy for the synthesis of 1-deoxy-azasugars from a chiral glycine equivalent and 4-carbon building blocks is described. Diastereoselective aldol additions of metalated bislactim ethers to matched and mismatched erythrose or threose acetonides and intramolecular N-alkylation (by reductive amination or nucleophilic substitution) were used as key steps. The dependence of the yield and the asymmetric induction of the aldol addition with the nature of the metallic counterion of the azaenolate and the γ-alkoxy protecting group for the erythrose or threose acetonides has been studied. The stereochemical outcome of the aldol additions with tin(II) azaenolates has been rationalized with the aid of density functional theory (DFT) calculations. In accordance with DFT calculations with model glyceraldehyde acetonides, hightrans,syn,anti-selectivitity for the matched pairs and moderate to low trans,anti,anti-selectivity for the mismatched ones may originate from (1) the intervention of solvated aggregates of tin(II) azaenolate and lithium chloride as the reactive species and (2) favored chair-like transition structures with a Cornforth-like conformation for the aldehyde moiety. DFT calculations indicate that aldol additions to erythrose acetonides proceed by an initial deprotonation, followed by coordination of the alkoxy-derivative to the tin(II) azaenolate and final reorganization of the intermediate complex through pericyclic transition structures in which the erythrose moiety is involved in a seven-membered chelate ring. The preparative utility of the aldol-based approach was demonstrated by application in concise routes for the synthesis of the glycosidase inhibitors 1-deoxy-d-allonojirimycin, 1-deoxy-l-altronojirimycin, 1-deoxy-d-gulonojirimycin, 1-deoxy-d-galactonojirimycin, 1-deoxy-l-idonojirimycin and 1-deoxy-d-talonojirimycin.
…………………..
http://pubs.acs.org/doi/abs/10.1021/jo00002a057
………………
Hinsken, Werner; DE 3906463 A1 1990
http://www.google.com/patents/DE3906463A1?cl=de
Example 1 Preparation of 1,5-dideoxy-1,5-imino-D-glucitol hydrobromide
A suspension of 1,5-dideoxy-1,5-imino-D-glucitol (500 g) in isopropanol (2 l) with 48% hydrochloric acid, bromine (620 g). The suspension is stirred for 2 hours at 40 ° C, cooled to 0 ° C and the product isolated by filtration.
Yield: 700 g (93% of theory),
mp: 184 ° C.
Example 2 Preparation of 1,5-dideoxy-1,5-imino-D-mannitol hydrobromide
The prepared analogously to Example 1 from 1,5-dideoxy 1,5-imino-D-mannitol and 48% hydrobromic acid.
Yield: 89% of theory;
C₆H₁₄NO₄Br (244.1)
Ber .: C 29.5%; H 5.8%; N 5.7%; Br 32.7%;
vascular .: C 29.8%; H 5.8%; N 5.8%; Br 32.3%.
Example 3 Preparation of 1,5-dideoxy-1,5-imino-D-Galactitol- hydrochloride
The preparation was carried out analogously to Example 1 from 1,5-dideoxy-1,5-imino-D-galactitol and corresponding mole ratios of 37% hydrochloric acid.
yield: 91% of theory
, mp: 160-162 ° C.
| Amat et al., “Eantioselective Synthesis of 1-deoxy-D-gluonojirimycin From A Phenylglycinol Derived Lactam,” Tetrahedron Letters, pp. 5355-5358, 2004. | ||
| 2 | Chernois, “Semimicro Experimental Organic Chemistry,” J. de Graff (1958), pp. 31-48. | |
| 3 | Encyclopedia of Chemical Technology, 4th Ed., 1995, John Wiley & Sons, vol. 14: p. 737-741. | |
| 4 | Heiker et al., “Synthesis of D-galacto-1-deoxynojirimycin (1, 5-dideoxy-1, 5-imino-D-galactitol) starting from 1-deoxynojirimycin.” Carbohydrate Research, 203: 314-318, 1990. | |
| 5 | Heiker et al., 1990, “Synthesis of D-galacto-1-deoxynojirimycin (1,5-dideoxy-1, 5-imino-D-galactitol) starting from 1-deoxynojirimycin,” Carbohydrate Research, vol. 203: p. 314-318. | |
| 6 | * | Joseph, Carbohydrate Research 337 (2002) 1083-1087. |
| 7 | * | Kinast et al. Angew. Chem. Int. Ed. Engl. 20 (1998), No. 9, pp. 805-806. |
| 8 | * | Lamb, Laboratory Manual of General Chemistry, Harvard University Press, 1916, p. 108. |
| 9 | Linden et al., “1-Deoxynojirimycin Hydrochloride,” Acta ChrystallographicaC50, pp. 746-749, 1994. | |
| 10 | Mellor et al., Preparation, biochemical characterization and biological properties of radiolabelled N-alkylated deoxynojirimycins, Biochem. J. Aug. 15, 2002; 366(Pt 1):225-233. | |
| 11 | * | Mills, Encyclopedia of Reagents for Organic Synthesis, Hydrochloric Acid, 2001 John Wily & Sons. |
| 12 | Santoyo-Gonzalez et al., “Use of N-Pivaloyl Imidazole as Protective Reagent for Sugars.” Synthesis 1998 1787-1792. | |
| 13 | Schuller et al., “Synthesis of 2-acetamido-1, 2-dideoxy-D-galacto-nojirimycin (2-acetamido-1, 2, 5-trideoxy-1, 5-imino-D-galacitol) from 1-deoxynojirimycin.” Carbohydrate Res. 1990; 203: 308-313. | |
| 14 | Supplementary European Search Report dated Mar. 11, 2010 issued in corresponding European Patent Application No. EP 06 77 2888. | |
| 15 | Uriel et al., A Short and Efficient Synthesis of 1,5-dideoxy-1,5-imino-D-galactitol (1-deoxy-D-galactostatin) and 1,5-dideoxy-1,5-dideoxy-1,5-imino-L-altritol (1-deoxy-L-altrostatin) From D-galactose, Synlett (1999), vol. 5, pp. 593-595. |
1-Deoxygalactonojirimycin:
(a) Liguchi, T.; Tajiri, K.; Ninomiya, I.; Naito, T. Tetrahedron2000, 56, 5819−5833.
(b) Mehta, G.; Mohal, N. Tetrahedron Lett. 2000, 41, 5741−5745.
(c) Asano, K.; Hakogi, T.; Iwama, S.; Katsumura, S. Chem. Commun. 1999, 41−42.
(d) Johnson, C. R.; Golebiowsky, A.; Sundram, H.; Miller, M. W.; Dwaihy, R. L. TetraherdonLett. 1995, 36, 653−654.
(e) Uriel, C.; Santoyo-Gonzalez, F. Synlett 1999, 593−595.
(f) Ruiz, M.; Ruanova, T. M.; Ojea, V.; Quintela, J. M. Tetrahedron Lett. 1999, 40, 2021−2024.
(g) Shilvock, J. P.; Fleet, G. W. J. Synlett 1998, 554−556.
(h) Chida, N.; Tanikawa, T.; Tobe, T.; Ogawa, S. J. Chem. Soc., Chem. Commun. 1994, 1247−1248.
(i) Aoyagi, S.; Fujimaki, S.; Yamazaki, N.; Kibayashi, C. J. Org. Chem. 1991, 56, 815−819.
(j) Kajimoto, T.; Chen, L.; Liu, K. K. C.; Wong, C. H. J. Am. Chem. Soc. 1991, 113, 6678−6680.
(k) Bernotas, R. C.; Pezzone, M. A.; Ganem, B. Carbohydr. Res. 1987, 167, 305−311. 1-Deoxyidonojirimycin:
(l) Singh, O. V.; Han, H. Tetrahedron Lett. 2003, 44, 2387−2391.
(m) Schaller, C.; Vogel, P.; Jager, V. Carbohydr. Res. 1998, 314, 25−35.
(n) Fowler, P. A.; Haines, A. H.; Taylor, R. J. K.; Chrystal, E. J. T.; Gravestock, M. B. Carbohydr. Res. 1993,246 377−381.
(o) Liu, K. K. C.; Kajimoto, T.; Chen, L.; Zhong, Z.; Ichikawa, Y.; Wong, C. H.J. Org. Chem. 1991, 56, 6280−6289. 1-Deoxygulonojirimycin: ref 5l.
(p) Haukaas, M. H.; O’Doherty, G. A. Org. Lett. 2001, 3, 401−404.
(q) Ruiz, M.; Ojea, V.; Ruanova, T. M.; Quintela, J. M. Tetrahedron: Asymmetry 2002, 13, 795−799. (r) Liao, L.-X.; Wang, Z.-M.; Zhang, H.-X.; Zhou, W.-S. Tetrahedron: Asymmetry 1999, 10, 3649−3657.
- See more at: http://worlddrugtracker.blogspot.in/#sthash.mFuiI6Hm.dpuf


Duvelisib
Infinity and AbbVie partner to develop and commercialise duvelisib for cancer
INK 1197; IPI 145; 8-Chloro-2-phenyl-3-[(1S)-1-(9H-purin-6-ylamino)ethyl]-1(2H)-isoquinolinone
1(2H)-Isoquinolinone, 8-chloro-2-phenyl-3-((1S)-1-(9H-purin-6-ylamino)ethyl)-
8-Chloro-2-phenyl-3-((1S)-1-(7H-purin-6-ylamino)ethyl)isoquinolin-1(2H)-one
(S)-3-(l-(9H-purin-6-ylamino)ethyl)-8-chloro-2-phenylisoquinolin-l(2H)-one
UNII-610V23S0JI; IPI-145; INK-1197;
Originator…….. Millennium Pharmaceuticals
| Molecular Formula | C22H17ClN6O | |
| Molecular Weight | 416.86 | |
| CAS Registry Number | 1201438-56-3 |

Infinity Pharmaceuticals has partnered with AbbVie to develop and commercialise its duvelisib (IPI-145), an oral inhibitor of phosphoinositide-3-kinase (PI3K)-delta and PI3K-gamma, to treat patients with cancer.
![]()
Infinity Pharmaceuticals has partnered with AbbVie to develop and commercialise its duvelisib (IPI-145), an oral inhibitor of phosphoinositide-3-kinase (PI3K)-delta and PI3K gamma, to treat patients with cancer.
Duvelisib has shown clinical activity against different blood cancers, such as indolent non-Hodgkin’s lymphoma (iNHL) and chronic lymphocytic leukemia (CLL).
AbbVie executive vice-president and chief scientific officer Michael Severino said: “We believe that duvelisib is a very promising investigational treatment based on clinical data showing activity in a broad range of blood cancers.”

Duvelisib (IPI-145, INK-1197), an inhibitor of PI3K-delta and –gamma, originated at Takeda subsidiary Intellikine. It is now being developed by Infinity Pharmaceuticals, which began a phase III trial in November, following US and EU grant of orphan drug status for both CLL and small lymphocytic leukemia
INK-1197 is a dual phosphatidylinositol 3-Kinase delta (PI3Kdelta) and gamma (PI3Kgamma) inhibitor in phase III clinical development at Infinity Pharmaceuticals for the treatment of chronic lymphocytic leukemia and small lymphocytic lymphoma. The company is also carring phase II trials for the treatment of patients with mild asthma undergoing allergen challenge, for the treatment of rheumatoid arthritis and for the treatment of refractory indolent non-Hodgkin’s lymphoma. Phase I clinical trials for the treatment of advanced hematological malignancies (including T-cell lymphoma and mantle cell lymphoma) are currently under way.
IPI-145 is an oral inhibitor of phosphoinositide-3-kinase (PI3K)-delta and PI3K-gamma. The PI3K-delta and PI3K-gamma isoforms are preferentially expressed in leukocytes (white blood cells), where they have distinct and non-overlapping roles in key cellular functions, including cell proliferation, cell differentiation, cell migration and immunity. Targeting PI3K-delta and PI3K-gamma may provide multiple opportunities to develop differentiated therapies for the treatment of blood cancers and inflammatory diseases.
Licensee Infinity Pharmaceuticals is developing INK-1197. In 2014, Infinity licensed Abbvie for joint commercialization in the U.S. and exclusive commercialization elsewhere. Originator Millennium Pharmaceuticals had also been developing the compound; however, no recent development has been reported for this research. In 2013, orphan drug designations were assigned by the FDA and the EMA for the treatment of chronic lymphocytic leukemia, for the treatment of small lymphocytic lymphoma and for the treatment of follicular lymphoma.
currently enrolling patients DYNAMO™, a Phase 2 study designed to evaluate the activity and safety of IPI-145 in approximately 120 people with refractory indolent non-Hodgkin lymphoma (iNHL) and DUO™, a Phase 3 clinical study of IPI-145 in approximately 300 people with relapsed/refractory chronic lymphocytic leukemia (CLL). These studies are supported by Phase 1 data reported at the 2013 American Society of Hematology (ASH) Annual Meeting which showed that IPI-145 was well tolerated and clinically active in a broad range of malignancies, including iNHL and CLL. These studies are part of DUETTS™, a worldwide investigation of IPI-145 in blood cancers.
WO 2011008302
http://www.google.com/patents/WO2011008302A1?cl=en
Reaction Scheme 1
Reaction Scheme 2:
201 202 203
204 205
Reaction Scheme 3:
Reaction Scheme 4A:
Reaction Scheme 4B:
2
Example 14b: Synthesis of (S)-3-(l-(9H-purin-6-ylamino)ethyl)-8-chloro-2-phenylisoquinolin-l(2H)-one (9)
(compound 4904)
Scheme 27b. The synthesis of (S)-3-(l-(9H-purin-6-ylamino)ethyl)-8-chloro-2-phenylisoquinolin-l(2H)-one (9)
(compound 4904) is described.
[00493] The compound of Formula 4904 (compound 292 in Table 4) was synthesized using the synthetic transformations as described in Examples 12 and 14a, but 2-chloro-6-methyl benzoic acid (compound 4903) was used instead of 2, 6 ,dimethyl benzoic acid (compound 4403). By a similar method, compound 328 in Table 4 was synthesized using the synthetic transformations as described starting from the 2-chloro-6-methyl m-fluorobenzoic acid.
…………………………………….
http://www.google.com/patents/WO2012097000A1?cl=en OR http://www.google.com/patents/US8809349?cl=en
Formula (I):
(I),
or a pharmaceutically acceptable salt, solvate, or hydrate thereof. In one embodiment, the method comprises any one, two, three, four, five, six, seven, or eight, or more of the following steps:
“Formula (I)” includes (S)-3-(l -(9H-purin-6-ylamino)ethyl)-8-chloro-2- phenylisoquinolin-l(2H)-one in its imide tautomer shown below as (1-1) and in its lactim tautomer shown below as (1-2):
(1-1)………………………………………………………………………………… (1-2)
[0055] FIG. 27 shows an FT-IR spectra of Polymorph Form C.

[0056] FIG. 28 shows a ‘H-NMR spectra of Polymorph Form C.

[0057] FIG. 29 shows a 13C-NMR spectra of Polymorph Form C.

Example 1
Synthesis of (S)-3-(l-aminoethyl)-8-chloro-2-phenylisoquinolin-l(2H)-one
Example 1A
1 2
[00563] Compound 1 (6.00 kg) was treated with 1-hydroxybenzotriazole monohydrate (HOBt»H20), triethylamine, Ν,Ο-dimethylhydroxylamine hydrochloride, and EDCI in dimethylacetamide (DMA) at
10 °C. The reaction was monitored by proton NMR and deemed complete after 2.6 hours, affording Compound 2 as a white solid in 95% yield. The R-enantiomer was not detected by proton NMR using (R)-(- ) -alpha-ace tylmandelic acid as a chiral-shift reagent.
[00564] Compound 3 (4.60 kg) was treated with p-toluenesulfonic acid monohydrate and 3,4-dihydro-2H- pyran (DHP) in ethyl acetate at 75 °C for 2.6 hours. The reaction was monitored by HPLC. Upon completion of the reaction, Compound 4 was obtained as a yellow solid in 80% yield with >99% (AUC) purity by HPLC analysis.
[00565] Compound 5 (3.30 kg) was treated with thionyl chloride and a catalytic amount of DMF in methylene chloride at 25 °C for five hours. The reaction was monitored by HPLC which indicated a 97.5% (AUC) conversion to compound 6. Compound 6 was treated in situ with aniline in methylene chloride at 25 °C for 15 hours. The reaction was monitored by HPLC and afforded Compound 7 as a brown solid in 81% yield with >99% (AUC) purity by HPLC analysis. [00566] Compound 2 was treated with 2.0 M isopropyl Grignard in THF at -20 °C. The resulting solution was added to Compound 7 (3.30 kg) pre -treated with 2.3 M n-hexyl lithium in tetrahydrofuran at -15 °C. The reaction was monitored by HPLC until a 99% (AUC) conversion to Compound 8 was observed.
Compound 8 was treated in situ with concentrated HC1 in isopropyl alcohol at 70 °C for eight hours. The reaction was monitored by HPLC and afforded Compound 9 as a brown solid in 85% yield with 98% (AUC) purity and 84% (AUC) ee by HPLC analysis.
Example ID
[00567] Compound 9 (3.40 kg) was treated with D-tartaric acid in methanol at 55 °C for 1-2 hours. The batch was filtered and treated with ammonium hydroxide in deionized (DI) water to afford enantiomerically enriched Compound 9 as a tan solid in 71% yield with >99% (AUC) purity and 91% (AUC) ee by HPLC analysis.
Example 2
Synthesis of (S)-3-(l-aminoethyl)-8-chloro-2-phenylisoquinolin-l(2H)-one
Example 2A
[00568] To Compound 7 (20.1 g) was charged 100 mL of anhydrous THF. The resulting solution was cooled to about -10 °C and 80 mL of n-hexyl lithium (2.3 M in hexanes, 2.26 equiv.) was slowly added (e.g. , over about 20 min). The resulting solution was stirred at about -10 °C for about 20 min.
[00569] To Compound 2 (26.5 g; 1.39 equiv.) was charged 120 mL of anhydrous THF. The resulting mixture was cooled to about -10 °C and 60 mL of isopropyl magnesium chloride (2.0 M in THF, 1.47 equiv.) was slowly added (e.g. , over about 15-20 min). The resulting mixture was then stirred at about -10 °C for about 20 min. The mixture prepared from Compound 2 was added to the solution prepared from Compound 7 while maintaining the internal temperature between about -10 and about 0 °C. After the addition was complete (about 5 min), the cold bath was removed, and the resulting mixture was stirred at ambient temperature for about 1 h, then cooled. [00570] A solution of 100 mL of anisole and 33 mL of isobutyric acid (4.37 equiv.) was prepared. The anisole solution was cooled to an internal temperature of about -3 °C. The above reaction mixture was added to the anisole solution such that the internal temperature of the anisole solution was maintained at below about 5 °C. The ice bath was then removed (after about 15 min, the internal temperature was about 7 °C). To the mixture, 100 mL of 10 wt aqueous NaCl solution was rapidly added (the internal temperature increased from about 7 °C to about 15 °C). After stirring for about 30 min, the two phases were separated. The organic phase was washed with another 100 mL of 10 wt aqueous NaCl. The organic phase was transferred to a flask using 25 mL of anisole to facilitate the transfer. The anisole solution was then concentrated to 109 g. Then, 100 mL of anisole was added.
[00571] To the approximately 200 mL of anisole solution was added 50 mL of TFA (8 equiv.) while maintaining the internal temperature below about 45-50 °C. The resulting solution warmed to about 45-50 °C and stirred for about 15 hrs, then cooled to 20-25 °C. To this solution was added 300 mL of MTBE dropwise and then the resulting mixture was held at 20-25 °C for 1 h. The mixture was filtered, and the wet cake washed with approximately 50 mL of MTBE. The wet cake was conditioned on the filter for about 1 h under nitrogen. The wet cake was periodically mixed and re-smoothed during conditioning. The wet cake was then washed with 200 mL of MTBE. The wet cake was further conditioned for about 2 h (the wet cake was mixed and resmoothed after about 1.5 h). The wet cake was dried in a vacuum oven at about 40 °C for about 18 h to afford Compound 9»TFA salt in about 97.3% purity (AUC), which had about 99.1 % S- enantiomer (e.g. , chiral purity of about 99.1 %).
[00572] Compound 9»TFA salt (3 g) was suspended in 30 mL of EtOAc at about 20 °C. To the EtOAc suspension was added 4.5 mL (2.2 eq.) of a 14% aqueous ammonium hydroxide solution and the internal temperature decreased to about 17 °C. Water (5 mL) was added to the biphasic mixture. The biphasic mixture was stirred for 30 min. The mixing was stopped and the phases were allowed to separate. The aqueous phase was removed. To the organic phase (combined with 5 mL of EtOAc) was added 10 mL of 10% aqueous NaCl. The biphasic mixture was stirred for about 30 min. The aqueous phase was removed. The organic layer was concentrated to 9 g. To this EtOAc mixture was added 20 mL of i-PrOAc. The resulting mixture was concentrated to 14.8 g. With stirring, 10 mL of n-heptane was added dropwise. The suspension was stirred for about 30 min, then an additional 10 mL of n-heptane was added. The resulting suspension was stirred for 1 h. The suspension was filtered and the wet cake was washed with additional heptane. The wet cake was conditioned for 20 min under nitrogen, then dried in a vacuum oven at about 40 °C to afford Compound 9 free base in about 99.3% purity (AUC), which had about 99.2% S-enantiomer (e.g., chiral purity of about 99.2%).
Example 2B [00573] A mixture of Compound 7 (100 g, 0.407 mol, 1 wt) and THF (500 mL, 5 vol) was prepared and cooled to about 3 °C. n-Hexyllithium (2.3 M in hexanes, 400 mL, 0.920 mol, 2.26 equiv) was charged over about 110 minutes while maintaining the temperature below about 6 °C. The resulting solution was stirred at 0 ± 5 °C for about 30 minutes. Concurrently, a mixture of Compound 2 (126 g, 0.541 mol, 1.33 equiv) and THF (575 mL, 5.8 vol) was prepared. The resulting slurry was charged with isopropylmagnesium chloride (2.0 M in THF, 290 mL, 0.574 mol, 1.41 equiv) over about 85 minutes while maintaining the temperature below about 5 °C. The resulting mixture was stirred for about 35 minutes at 0 ± 5 °C. The Compound 2 magnesium salt mixture was transferred to the Compound 7 lithium salt mixture over about 1 hour while maintaining a temperature of 0 ± 5 °C. The solution was stirred for about 6 minutes upon completion of the transfer.
[00574] The solution was added to an about -5 °C stirring solution of isobutyric acid (165 mL, 1.78 mol, 4.37 equiv) in anisole (500 mL, 5 vol) over about 20 minutes during which time the temperature did not exceed about 6 °C. The resulting solution was stirred for about 40 minutes while being warmed to about 14 °C. Then, a 10% sodium chloride solution (500 mL, 5 vol) was rapidly added to the reaction. The temperature rose to about 21 °C. After agitating the mixture for about 6 minutes, the stirring was ceased and the lower aqueous layer was removed (about 700 mL). A second portion of 10% sodium chloride solution (500 mL, 5 vol) was added and the mixture was stirred for 5 minutes. Then, the stirring was ceased and the lower aqueous layer was removed. The volume of the organic layer was reduced by vacuum distillation to about 750 mL (7.5 vol).
[00575] Trifluoroacetic acid (250 mL, 3.26 mol, 8.0 equiv) was added and the resulting mixture was agitated at about 45 °C for about 15 hours. The mixture was cooled to about 35 °C and MTBE (1.5 L, 15 vol) was added over about 70 minutes. Upon completion of the addition, the mixture was agitated for about 45 minutes at about 25-30 °C. The solids were collected by vacuum filtration and conditioned under N2 for about 20 hours to afford Compound 9*TFA salt in about 97.5% purity (AUC), which had a chiral purity of about 99.3%.
[00576] Compound 9»TFA salt (100 g) was suspended EtOAc (1 L,10 vol) and 14% aqueous ammonia (250 mL, 2.5 vol). The mixture was agitated for about 30 minutes, then the lower aqueous layer was removed. A second portion of 14% aqueous ammonia (250 mL, 2.5 vol) was added to the organic layer. The mixture was stirred for 30 minutes, then the lower aqueous layer was removed. Isopropyl acetate (300 mL, 3 vol) was added, and the mixture was distilled under vacuum to 500 mL (5 vol) while periodically adding in additional isopropyl acetate (1 L, 10 vol).
[00577] Then, after vacuum-distilling to a volume of 600 mL (6 vol), heptanes (1.5 L, 15 vol) were added over about 110 minutes while maintaining a temperature between about 20 °C and about 30 °C. The resulting slurry was stirred for about 1 hour, then the solid was collected by vacuum filtration. The cake was washed with heptanes (330 mL, 3.3 vol) and conditioned for about 1 hour. The solid was dried in an about 45 °C vacuum oven for about 20 hours to afford Compound 9 free base in about 99.23% purity (AUC), which has a chiral purity of about 99.4%.
Example 3
Chiral Resolution of (S)-3-(l-aminoethyl)-8-chloro-2-phenylisoquinolin-l(2H)-one (Compound 9)
[00578] In some instances, (S)-3-(l-aminoethyl)-8-chloro-2-phenylisoquinolin-l(2H)-one (Compound 9) obtained by synthesis contained a minor amount of the corresponding (R)-isomer. Chiral resolution procedures were utilized to improve the enantiomeric purity of certain samples of (S)-3-(l-aminoethyl)-8- chloro-2-phenylisoquinolin- 1 (2H)-one.
[00579] In one experiment, Compound 9 (3.40 kg) was treated with D-tartaric acid in methanol at about 55 °C for about 1 to about 2 hours. The mixture was filtered and treated with ammonium hydroxide in deionized (DI) water to afford Compound 9 in greater than about 99% (AUC) purity, which had a chiral purity of about 91% (AUC).
[00580] In another procedure, MeOH (10 vol.) and Compound 9 (1 equiv.) were stirred at 55 ± 5 °C. D- Tartaric acid (0.95 equiv.) was charged. The mixture was held at 55 ± 5 °C for about 30 min and then cooled to about 20 to about 25 °C over about 3 h. The mixture was held for about 30 min and then filtered. The filter cake was washed with MeOH (2.5 vol.) and then conditioned. The cake was returned to the reactor and water (16 vol.) was charged. The mixture was stirred at 25 ± 5 °C. NH4OH was then charged over about 1 h adjusting the pH to about 8 to about 9. The mixture was then filtered and the cake was washed with water (4 vol.) and then heptanes (4 vol.). The cake was conditioned and then vacuum dried at 45-50 °C to afford Compound 9 free base with a chiral purity of about 99.0%.
Example 4
Synthesis of (S)-3-(l-(9H-purin-6-ylamino)ethyl)-8-chloro-2-phenylisoquinolin-l(2H)-one
[00581] A mixture of Compound 7 (1 equiv.) and anhydrous THF (5 vol.) was prepared. Separately, a mixture of Compound 2 (1.3 equiv.) and anhydrous THF (5 vol.) was prepared. Both mixtures were stirred for about 15 min at about 20 to about 25 °C and then cooled to -25 ± 15 °C. n-Hexyl lithium (2.05 equiv.) was added to the Compound 7 mixture, maintaining the temperature at > 5 °C. i-PrMgCl (1.33 equiv.) was added to the Compound 2 mixture, maintaining the temperature at > 5 °C. The Compound 2 mixture was transferred to the Compound 7 mixture under anhydrous conditions at 0 ± 5 °C. The resulting mixture was warmed to 20 ± 2 °C and held for about 1 h. Then, the reaction was cooled to -5 ± 5 °C, and 6 N HC1 (3.5 equiv.) was added to quench the reaction, maintaining temperature at below about 25 °C. The aqueous layer was drained, and the organic layer was distilled under reduced pressure until the volume was 2-3 volumes. IPA (3 vol.) was added and vacuum distillation was continued until the volume was 2-3 volumes. IPA (8 vol.) was added and the mixture temperature was adjusted to about 60 °C to about 75 °C. Cone. HC1 (1.5 vol.) was added and the mixture was subsequently held for 4 hours. The mixture was distilled under reduced pressure until the volume was 2.5-3.5 volumes. The mixture temperature was adjusted to 30 ± 10 °C. DI water (3 vol.) and DCM (7 vol.) were respectively added to the mixture. Then, NH4OH was added to the mixture, adjusting the pH to about 7.5 to about 9. The temperature was adjusted to about 20 to about 25 °C. The layers were separated and the aqueous layer was washed with DCM (0.3 vol.). The combined DCM layers were distilled until the volume was 2 volumes. i-PrOAc (3 vol.) was added and vacuum distillation was continued until the volume was 3 volumes. The temperature was adjusted to about 15 to about 30 °C. Heptane (12 vol.) was charged to the organic layer, and the mixture was held for 30 min. The mixture was filtered and filter cake was washed with heptane (3 vol.). The cake was vacuum dried at about 45 °C afford Compound 9.
[00582] Then, MeOH (10 vol.) and Compound 9 (1 equiv.) were combined and stirred while the temperature was adjusted to 55 ± 5 °C. D-Tartaric acid (0.95 equiv.) was charged. The mixture was held at 55 ± 5 °C for about 30 min and then cooled to about 20 to about 25 °C over about 3 h. The mixture was held for 30 min and then filtered. The filter cake was washed with MeOH (2.5 vol.) and then conditioned. Water (16 vol.) was added to the cake and the mixture was stirred at 25 ± 5 °C. NH4OH was charged over 1 h adjusting the pH to about 8 to about 9. The mixture was then filtered and the resulting cake washed with water (4 vol.) and then heptanes (4 vol.). The cake was conditioned and then vacuum dried at 45-50 °C to afford Compound 9.
[00583] To a mixture of i-PrOH (4 vol.) and Compound 9 (1 equiv.) was added Compound 4 (1.8 equiv.), Et3N (2.5 equiv.) and i-PrOH (4 vol.). The mixture was agitated and the temperature was adjusted to 82 ± 5 °C. The mixture was held for 24 h. Then the mixture was cooled to about 20 to about 25 °C over about 2 h. The mixture was filtered and the cake was washed with i-PrOH (2 vol.), DI water (25 vol.) and n-heptane (2 vol.) respectively. The cake was conditioned and then vacuum dried at 50 ± 5 °C to afford Compound 10.
To a mixture of EtOH (2.5 vol.) and Compound 10 (1 equiv.) was added EtOH (2.5 vol.) and DI water (2 vol.). The mixture was agitated at about 20 to about 25 °C. Cone. HC1 (3.5 equiv.) was added and the temperature was adjusted to 35 ± 5 °C. The mixture was held for about 1.5 h. The mixture was cooled to 25 ± 5 °C and then polish filtered to a particulate free vessel. NH4OH was added, adjusting the pH to about 8 to about 9. Crystal seeds of Form C of a compound of Formula (I) (0.3 wt ) were added to the mixture which was held for 30 minutes. DI water (13 vol.) was added over about 2 h. The mixture was held for 1 h and then filtered. The resulting cake was washed with DI water (4 vol.) and n-heptane (2 vol.) respectively. The cake was conditioned for about 24 h and then DCM (5 vol.) was added. This mixture was agitated for about 12 h at about 20 to about 25 °C. The mixture was filtered and the cake washed with DCM (1 vol.). The cake was conditioned for about 6 h. The cake was then vacuum-dried at 50 ± 5 °C. To the cake was added DI water (10 vol.), and i-PrOH (0.8 vol.) and the mixture was agitated at 25 ± 5 °C for about 6 h. An XRPD sample confirmed the compound of Formula (I) was Form C. The mixture was filtered and the cake was washed with DI water (5 vol.) followed by n-heptane (3 vol.). The cake was conditioned and then vacuum dried at 50 ± 5 °C to afford a compound of Formula (I) as polymorph Form C. Example 5
Synthesis of (S)-3-(l-(9H-purin-6-ylamino)ethyl)-8-chloro-2-phenylisoquinolin-l(2H)-one
Example 5A
[00584] Compound 9 (2.39 kg) was treated with Compound 4 and triethylamine in isopropyl alcohol at 80 °C for 24 hours. The reaction was monitored by HPLC until completion, affording 8-chloro-2-phenyl-3- ((lS)-l-(9-(tetrahydro-2H^yran-2-yl)-9H^urin-6-ylamino)ethyl)isoquinolin-l(2H)-one (compound 10) as a tan solid in 94% yield with 98% (AUC) purity by HPLC analysis.
[00585] 8-Chloro-2-phenyl-3-((lS)-l-(9-(tetrahydro-2H-pyran-2-yl)-9H-purin-6-ylamino)ethyl)- isoquinolin-l(2H)-one (compound 10) (3.63 kg) was treated with HC1 in ethanol at 30 °C for 2.3 hours. The reaction was monitored by HPLC until completion, and afforded a compound of Formula (I) as a tan solid in 92% yield with >99% (AUC) purity and 90.9% (AUC) ee by HPLC analysis.
Example 5B
[00586] 3-(l-Aminoethyl)-8-chloro-2-phenylisoquinolin-l(2H)-one (Compound 9) (0.72 mmol), 6-chloro- 9-(tetrahydro-2H-pyran-2-yl)-9H-purine (Compound 4) (344 mg, 1.44 mmol) and DIPEA
(279 mg, 2.16 mmol) were dissolved in «-BuOH (20 mL), and the resulting mixture was stirred at reflux for 16 h. The reaction mixture was concentrated in vacuo and purified by flash column chromatography on silica gel (eluting with 30% to 50% Hex/EA) to afford the product, 8-chloro-2-phenyl-3-((lS)-l-(9-(tetrahydro-2H- pyran-2-yl)-9H-purin-6-ylamino)ethyl)isoquinolin-l(2H)-one (Compound 10), as a white solid (60% yield). [00587] 8-Chloro-2-phenyl-3-((lS)-l-(9-(tetrahydro-2H-pyran-2-yl)-9H-purin-6-ylamino)ethyl)- isoquinolin-l(2H)-one (Compound 10) (0.42 mmol) was dissolved in HCl/EtOH (3 M, 5 mL), and the resulting mixture was stirred at room temperature for 1 h. The reaction mixture was quenched with saturated NaHC03 aqueous solution and the pH was adjusted to about 7-8. The mixture was extracted with CH2C12 (50 mL x 3), dried over anhydrous Na2S04, and filtered. The filtrate was concentrated in vacuo, and the residue was recrystallized from ethyl acetate and hexanes (1 : 1). The solid was collected by filtration and dried in vacuo to afford the product (S)-3-(l-(9H-purin-6-ylamino) ethyl)-8-chloro-2-phenylisoquinolin- l(2H)-one (Formula (I)) (90% yield) as a white solid as polymorph Form A.
Example 5C
[00588] 3-(l-Aminoethyl)-8-chloro-2-phenylisoquinolin-l(2H)-one (Compound 9) and 6-chloro-9- (tetrahydro-2H-pyran-2-yl)-9H-purine (Compound 4) are combined in the presence of triethylamine and isopropyl alcohol. The reaction solution is heated at 82 °C for 24 hours to afford Compound 10. The intermediate compound 10 is treated with concentrated HCl and ethanol under aqueous conditions at 35 °C to remove the tetrahydropyranyl group to yield (S)-3-(l-(9H-purin-6-ylamino)ethyl)-8-chloro-2- phenylisoquinolin-l(2H)-one. Isolation/purification under aqueous conditions affords polymorph Form C.
Example 6
Synthesis of (S)-3-(l-(9H^urin-6-ylamino)ethyl)-8-chloro-2-phenylisoquinolin-l(2H)-one
[00589] 3-(l-Aminoethyl)-8-chloro-2-phenylisoquinolin-l(2H)-one (Compound 9) (150 g; 90% ee) and 6- chloro-9-(tetrahydro-2H-pyran-2-yl)-9H-purine (Compound 4) (216 g, 1.8 equiv) were charged to a round bottom flask followed by addition of IPA (1.2 L; 8 vol) and triethylamine (175 mL; 2.5 equiv). The resultant slurry was stirred at reflux for one day. Heptane (1.5 L; 10 vol) was added dropwise over two hours. The batch was then cooled to 0-5 °C, held for one hour and filtered. The cake was washed with heptane (450 mL; 3 vol) and returned to the reactor. IPA (300 mL; 2 vol) and water (2.25 L; 15 vol) were added and the resultant slurry stirred at 20-25 °C for three and half hours then filtered. The cake was washed with water (1.5 L; 10 vol) and heptane (450 mL; 3 vol) and then vacuum dried at 48 °C for two and half days to give 227 g (90.1 %) of the intermediate (Compound 10) as an off-white solid with >99% (AUC) purity and >94 ee (chiral HPLC). The ee was determined by converting a sample of the cake to the final product and analyzing it with chiral HPLC.
[00590] The intermediate (Compound 10) (200 g) was slurried in an ethanol (900 mL; 4.5 vol) / water (300 mL; 1.5 vol) mixture at 22 °C followed by addition of cone. HC1 (300 mL; 1.5 vol) and holding for one and half hours at 25-35 °C. Addition of HC1 resulted in complete dissolution of all solids producing a dark brown solution. Ammonium hydroxide (260 mL) was added adjusting the pH to 8-9. Product seeds of polymorph Form C (0.5 g) (Form A seeds can also be used) were then added and the batch which was held for ten minutes followed by addition of water (3 L; 15 vol) over two hours resulting in crystallization of the product. The batch was held for 3.5 hours at 20-25 °C and then filtered. The cake was washed with water (1 L; 5 vol) followed by heptane (800 mL; 4 vol) and vacuum dried at 52 °C for 23 hours to give 155.5 g (93.5%) of product with 99.6% (AUC) purity and 93.8% ee (chiral HPLC).
Example 7
-3-(l-(9H-purin-6-ylamino)ethyl)-8-chloro-2-phenylisoquinolin-l(2H)-one
[00591] A mixtue of isopropanol (20.20 kg, 8 vol.), Compound 9 (3.17 kg, 9.04 mol, 1 eq.), Compound 4 (4.61 kg, 16.27 mol, 1.8 eq.) and triethylamine (2.62 kg, 20.02 mol, 2.4 eq.) was prepared and heated to an internal temperature of 82 ± 5 °C. The mixture was stirred at that temperature for an additional about 24 h. The temperature was adjusted to 20 ± 5 °C slowly over a period of about 2 h and the solids were isolated via vacuum filtration through a 24″ polypropylene table top filter equipped with a Sharkskin paper. The filter cake was rinsed sequentially with IPA (5.15 kg, 3 vol.), purified water (80.80 kg, 25 vol.) and n-heptane (4.30 kg, 2 vol.). The cake was further dried for about 4 days in vacuo at 50 ± 5 °C to afford Compound 10.
[00592] To a mixture of ethanol (17.7 kg, 5 vol.) and Compound 10 (4.45 kg, 8.88 mol. 1.0 eq.) was added purified water (8.94 kg, 2 vol.). To this mixture was slowly added concentrated HC1 (3.10 kg, 3.5 eq.) while maintaining the temperature below about 35 °C. The mixture was stirred at 30 ± 5 °C for about 1.5 h and HPLC analysis indicated the presence the compound of Formula (I) in 99.8% (AUC) purity with respect to compound 10.
[00593] Then, the compound of Formula (I) mixture was cooled to 25 ± 5 °C. The pH of the mixture was adjusted to about 8 using pre filtered ammonium hydroxide (1.90 kg). After stirring for about 15 min, Form C crystal seeds (13.88 g) were added. After stirring for about 15 min, purified water (58.0 kg, 13 vol.) was charged over a period of about 2 h. After stirring the mixture for 15 h at 25 ± 5 °C, the solids were isolated via vacuum filtration through a 24″ polypropylene table top filter equipped with a PTFE cloth over Sharkskin paper. The filter cake was rinsed with purified water (18.55 kg, 4 vol.) followed by pre -filtered n-heptane (6.10 kg, 2 vol.). After conditioning the filter cake for about 24 h, HPLC analysis of the filter cake indicated the presence the compound of Formula (I) in about 99.2% (AUC) purity.
[00594] To the filter cake was added dichloromethane (29.9 kg, 5 vol.) and the slurry was stirred at 25 ± 5 °C for about 24 h. The solids were isolated via vacuum filtration through a 24″ polypropylene table top filter equipped with a PTFE cloth over Sharkskin paper, and the filter cake was rinsed with DCM (6.10 kg, 1 vol.). After conditioning the filter cake for about 22 h, the filter cake was dried for about 2 days in vacuo at 50 ± 5 °C to afford the compound of Formula (I) in 99.6% (AUC) purity. The compound of Formula (I) was consistent with a Form A reference by XRPD.
[00595] To this solid was added purified water (44.6 kg, 10 vol.) and pre filtered 2-propanol (3.0 kg, 0.8 vol.). After stirring for about 6 h, a sample of the solids in the slurry was analyzed by XRPD and was consistent with a Form C reference. The solids were isolated via vacuum filtration through a 24″ polypropylene table top filter equipped with a PTFE cloth over Sharkskin paper, and the filter cake was rinsed with purified water (22.35 kg, 5 vol.) followed by pre filtered n-heptane (9.15 kg, 3 vol.). After conditioning the filter cake for about 18 h, the filter cake was dried in vacuo for about 5 days at 50 ± 5 °C.
[00596] This process afforded a compound of Formula (I) in about 99.6% (AUC) purity, and a chiral purity of greater than about 99% (AUC). An XRPD of the solid was consistent with a Form C reference standard. :H NMR (DMSO-<i6) and IR of the product conformed with reference standard.
……………………………………………


KEY Duvelisib, IPI-145, INK-1197, AbbVie, INFINITY, chronic lymphocytic leukemia, phase 3, orphan drug
| WO2013088404A1 | Dec 14, 2012 | Jun 20, 2013 | Novartis Ag | Use of inhibitors of the activity or function of PI3K |
| WO2014004470A1 * | Jun 25, 2013 | Jan 3, 2014 | Infinity Pharmaceuticals, Inc. | Treatment of lupus, fibrotic conditions, and inflammatory myopathies and other disorders using pi3 kinase inhibitors |
| WO2014072937A1 | Nov 7, 2013 | May 15, 2014 | Rhizen Pharmaceuticals Sa | Pharmaceutical compositions containing a pde4 inhibitor and a pi3 delta or dual pi3 delta-gamma kinase inhibitor |
| US7449477 * | Nov 22, 2004 | Nov 11, 2008 | Eli Lilly And Company | 7-phenyl-isoquinoline-5-sulfonylamino derivatives as inhibitors of akt (protein kinase B) |
| US20090312319 * | Jul 15, 2009 | Dec 17, 2009 | Intellikine | Certain chemical entities, compositions and methods |
| US20100168153 * | Nov 16, 2007 | Jul 1, 2010 | Novartis Ag | Salts and crystall forms of 2-methyl-2-[4-(3-methyl-2-oxo-8-quinolin-3-yl-2,3-dihydro-imidazo[4,5-c]quinolin-1-yl)-phenyl]-propionitrile |
| WO2013012915A1 | Jul 18, 2012 | Jan 24, 2013 | Infinity Pharmaceuticals Inc. | Heterocyclic compounds and uses thereof |
| WO2013012918A1 | Jul 18, 2012 | Jan 24, 2013 | Infinity Pharmaceuticals Inc. | Heterocyclic compounds and uses thereof |
| WO2013032591A1 | Jul 18, 2012 | Mar 7, 2013 | Infinity Pharmaceuticals Inc. | Heterocyclic compounds and uses thereof |
| WO2013049332A1 | Sep 27, 2012 | Apr 4, 2013 | Infinity Pharmaceuticals, Inc. | Inhibitors of monoacylglycerol lipase and methods of their use |
| WO2013088404A1 | Dec 14, 2012 | Jun 20, 2013 | Novartis Ag | Use of inhibitors of the activity or function of PI3K |
| WO2013154878A1 | Apr 3, 2013 | Oct 17, 2013 | Infinity Pharmaceuticals, Inc. | Heterocyclic compounds and uses thereof |
| WO2014004470A1 * | Jun 25, 2013 | Jan 3, 2014 | Infinity Pharmaceuticals, Inc. | Treatment of lupus, fibrotic conditions, and inflammatory myopathies and other disorders using pi3 kinase inhibitors |
| WO2014071105A1 | Nov 1, 2013 | May 8, 2014 | Infinity Pharmaceuticals, Inc. | Treatment of rheumatoid arthritis and asthma using p13 kinase inhibitors |
| WO2014071109A1 | Nov 1, 2013 | May 8, 2014 | Infinity Pharmaceuticals, Inc. | Treatment of cancers using pi3 kinase isoform modulators |
| WO2014072937A1 | Nov 7, 2013 | May 15, 2014 | Rhizen Pharmaceuticals Sa | Pharmaceutical compositions containing a pde4 inhibitor and a pi3 delta or dual pi3 delta-gamma kinase inhibitor |
| WO2001081346A2 | Apr 24, 2001 | Nov 1, 2001 | Icos Corp | Inhibitors of human phosphatidyl-inositol 3-kinase delta |
| US6800620 | Jan 6, 2003 | Oct 5, 2004 | Icos | Contacting leukocytes, osteoclasts with an enzyme inhibitors, a 9h-purin-3h-quinazolin-4-one derivatives, treating bone-resorption disorder, antiproliferative agents treating leukemia cells |
| US20060276470 * | Aug 18, 2003 | Dec 7, 2006 | Jackson Shaun P | (+-)-7-Methyl-2-morpholin-4-yl-9-(1-phenylaminoethyl)-pyrido[1,2-a]pyrimidin-4-one, for example; selective inhibitors of phosphoinositide (PI) 3-kinase beta for use in anti-thrombotic therapy |
| US20080032960 * | Apr 4, 2007 | Feb 7, 2008 | Regents Of The University Of California | PI3 kinase antagonists |

NINTEDANIB, BBIF 1120, Intedanib
Boehringer Ingelheim Corp
| chinese, japanese | 尼达尼布 ニンテダニブ |
Nintedanib (formerly BIBF 1120) is a small molecule tyrosine-kinase inhibitor, targeting vascular endothelial growth factor receptor (VEGFR), fibroblast growth factor receptor (FGFR) and platelet derived growth factor receptor (PDGFR) being developed by Boehringer Ingelheim as an anti-angiogenesis anti-cancer agent under the trade name Vargatef, and recently approved for treatment of idiopathic pulmonary fibrosis as Ofev.
Nintedanib is an indolinone-derived drug that inhibits the process of blood vessel formation (angiogenesis). Angiogenesis inhibitors stop the formation and reshaping of blood vessels in and around tumours, which reduces the tumour’s blood supply, starving tumour cells of oxygen and nutrients leading to cell death and tumour shrinkage. Unlike conventional anti-cancer chemotherapy which has a direct cell killing effect on cancer cells, angiogenesis inhibitors starve the tumour cells of oxygen and nutrients which results in tumour cell death. One of the advantages of this method of anti-cancer therapy is that it is more specific than conventional chemotherapy agents, therefore results in fewer and less severe side effects than conventional chemotherapy.
The process of new blood vessel formation (angiogenesis) is essential for the growth and spread of cancers. It is mediated by signaling molecules (growth factors) released from cancer cells in response to low oxygen levels. The growth factors cause the cells of the tumour’s blood vessel to divide and reorganize resulting in the sprouting of new vessels in and around the tumour, improving its blood supply.
Angiogenesis is a process that is essential for the growth and spread of all solid tumours, blocking it prevents the tumour from growing and may result in tumour shrinkage as well as a reduction in the spread of the cancer to other parts of the body. Nintedanib exerts its anti-cancer effect by binding to and blocking the activation of cell receptors involved in blood vessel formation and reshaping (i.e. VEGFR 1-3, FGFR 1-3 AND PDGFRα and β). Inhibition of these receptors in the cells that make up blood vessels (endothelial cells, smooth muscle cells and pericytes) by Nintedanib leads to programmed cell death, destruction of tumor blood vessels and a reduction in blood flow to the tumour. Reduced tumour blood flow inhibits tumor cell proliferation and migration hence slowing the growth and spread of the cancer.[1]
Preclinical studies have shown that nintedanib binds in a highly selective manner to the ATP binding pocked of its three target receptor families, without binding to similarly shaped ATP domains in other proteins, which reduces the potential for undesirable side effects.[2]
The most common side effects observed with nintedanib were reversible elevation in liver enzymes (10-28% of patients) and gastrointestinal disturbance (up to 50%). Side effects observed with nintedanib were worse with the higher 250 mg dose, for this reason subsequent trials have used the equally clinically effective 200 mg dose.[1][2][3][4][5][6][7][8][9]
Nintedanib inhibits the growth and reshaping of blood vessels which is also an essential process in normal wound healing and tissue repair. Therefore a theoretical side effect of nintedanib is reduced wound healing however, unlike other anti-angiogenic agents, this side effect has not been observed in patients receiving nintedanib.
Preclinical studies have demonstrated that nintedanib selectively binds to and blocks the VEGF, FGF and PDGF receptors, inhibiting the growth of cells that constitute the walls of blood vessels (endothelial and smooth muscle cells and pericytes) in vitro. Nintedanib reduces the number and density of blood vessels in tumours in vivo, resulting in tumour shrinkage.[1][2] Nintedanib also inhibits the growth of cells that are resistant to existing chemotherapy agents in vitro, which suggests a potential role for the agent in patients with solid tumours that are unresponsive to or relapse following current first line therapy.[10]
Early clinical trials of nintedanib have been carried out in patients with non-small cell lung, colorectal, uterine, endometrial, ovarian and cervical cancer and multiple myeloma.[4][5][7][8][9] These studies reported that the drug is active in patients, safe to administer and is stable in the bloodstream. They identified that the maximum tolerated dose of nintedanib is 20 0 mg when taken once a day.
In the first human trials, nintedanib halted the growth of tumours in up to 50% of patients with non-small cell lung cancer and 76% of patients with advanced colorectal cancer and other solid tumours.[4][8] A complete response was observed in 1/26 patients with non-small cell lung and 1/7 patients with ovarian cancer treated with nintedanib. A further 2 patients with ovarian cancer had partial responses to nintedanib.[8][9]
Two phase II trials have been carried out assessing the efficacy, dosing and side effects of nintedanib in non-small cell lung and ovarian cancer. These trials found that nintedanib delayed relapse in patients with ovarian cancer by two months[6] and that overall survival of patients with non-small cell lung who received nintedanib was similar to that observed with the FDA approved VEGFR inhibitor sorafenib. These trials also concluded that increasing the dose of the nintedanib has no effect on survival.[3]
Nintedanib is being tested in several phase I to III clinical trials for cancer. Angiogenesis inhibitors such as nintedanib may be effective in a range of solid tumour types including; lung, ovarian, metastatic bowel, liver and brain cancer. Patients are also being recruited for three phase III clinical trials that will evaluate the potential benefit of nintedanib when added to existing 1st line treatments in patients with ovarian.[11] and 2nd line treatment in non-small cell lung cancer [12][13] The phase III trials of nintedanib in lung cancer have been named LUME-Lung 1 and LUME-Lung 2.
Current phase II trials are investigating the effect of nintedanib in patients with metastatic bowel cancer, liver cancer and the brain tumour: glioblastoma multiforme.[14]
Phase III trials are investigating the use of nintedanib in combination with the existing chemotherapy agents permexetred and docetaxel in patients with non-small cell lung cancer,[15] and in combination with carboplatin and paclitaxel as a first line treatment for patients with ovarian cancer.[16]
A phase III clinical trial was underway examining the safety and efficacy of nintedanib on patients with the non-cancerous lung condition idiopathic pulmonary fibrosis.[17] Nintedanib, under the brand name Ofev, was approved by the FDA for treatment of idiopathic pulmonary fibrosis on 15 Oct 2014. [18]
In terms of clinical development, additional phase III clinical trials are ongoing for the treatment of epithelial ovarian cancer, fallopian tube or primary peritoneal cancer, in combination with chemotherapy, and for the treatment of refractory metastatic colorectal cancer. Phase II clinical trials are also ongoing at the company for the treatment of glioblastoma multiforme, previously untreated patients with renal cell cancer, and for the treatment of patients with unresectable malignant pleural mesothelioma. The National Cancer Center of Korea (NCC) is evaluating the compound in phase II studies as second line treatment for small cell lung cancer (SCLC). The Centre Oscar Lambret is also conducting phase II clinical trials for the treatment of breast cancer in combination with docetaxel. Phase II trials are under way at EORTC as second line therapy for patients with either differentiated or medullary thyroid cancer progressing after first line therapy. The compound is also in early clinical development for the treatment of cancer of the peritoneal cavity, hepatocellular carcinoma, acute myeloid leukemia and ovarian cancer. Clinical trials have been completed for the treatment of prostate cancer and for the treatment of colorectal cancer. Boehringer Ingelheim is also conducting phase I/II clinical trials for the treatment of NSCLC and acute myeloid leukemia in addition to low-dose cytarabine. Phase I clinical studies are ongoing at the company for the treatment of epithelial ovary cancer and for the treatment of patients with mild and moderate hepatic impairment. The company had been evaluating the compound in early clinical trials for the treatment of prostate cancer in combination with docetaxel, but recent progress reports for this indication are not available at present.
In 2011, orphan drug designation was assigned in the U.S. and Japan for the treatment of idiopathic pulmonary fibrosis. In 2013, orphan drug designation was also assigned for the same indication in the E.U. In 2014, a Breakthrough Therapy Designation was assigned to the compound for the treatment of idiopathic pulmonary fibrosis.
The present invention relates to a beneficial treatment of tumours in patients suffering from NSCLC, and to a clinical marker useful as predictive variable of the responsiveness of tumours in patients suffering from NSCLC. The present invention further relates to a method for selecting patients likely to respond to a given therapy, wherein said method optionally comprises the use of a specific clinical marker. The present invention further relates to a method for delaying disease progression and/or prolonging patient survival of NSCLC patients, wherein said method comprises the use of a specific clinical marker.
The monoethanesulphonate salt form of this compound presents properties which makes this salt form especially suitable for development as medicament. The chemical structure of 3-Z-[l-(4-(N-((4-methyl-piperazin-l-yl)-methylcarbonyl)-N-methyl-amino)-anilino)- 1 -phenyl-methylene] -6-methoxycarbonyl-2-indolinone-monoethanesulphonate (ΓΝΝ name nintedanib esylate) is depicted below as Formula Al .
Formula Al

This compound is thus for example suitable for the treatment of diseases in which angiogenesis or the proliferation of cells is involved. The use of this compound for the treatment of immunologic diseases or pathological conditions involving an
immunologic component is being described in WO 2004/017948, the use for the treatment of, amongst others, oncological diseases, alone or in combination, is being described in WO 2004/096224 and WO 2009/147218, and the use for the treatment of fibrotic diseases is being described in WO 2006/067165.
A method using biomarkers for monitoring the treatment of an individual with the compound 3-Z-[l-(4-(N-((4-methyl-piperazin-l-yl)-methylcarbonyl)-N-methyl-amino)-anilino)-l -phenyl-methylene] -6-methoxycarbonyl-2-indolinone or a pharmaceutically acceptable salt thereof, wherein it is determined if a sample from said individual comprises a biomarker in an amount that is indicative for said treatment, is disclosed in WO 2010/103058.
……………………………………………………………………………………………
http://www.google.com/patents/US20110201812
The present invention relates to a process for the manufacture of a specific indolinone derivative and a pharmaceutically acceptable salt thereof, namely 3-Z-[1-(4-(N-((4-methyl-piperazin-1-yl)-methylcarbonyl)-N-methyl-amino)-anilino)-1-phenyl-methylene]-6-methoxycarbonyl-2-indolinone and its monoethanesulfonate, to new manufacturing steps and to new intermediates of this process.
The indolinone derivative 3-Z-[1-(4-(N-((4-methyl-piperazin-1-yl)-methylcarbonyl)-N-methyl-amino)-anilino)-1-phenyl-methylene]-6-methoxycarbonyl-2-indolinone and its monoethanesulfonate are known from the following patent applications: WO 01/027081, WO 04/013099, WO 04/017948, WO 04/096224 and WO 06/067165. These patent applications disclose the compound, a process for its manufacture, a specific salt form of this compound and the use of the compound or its salt in a pharmaceutical composition to treat oncological or non-oncological diseases via inhibition of the proliferation of target cells, alone or in combination with further therapeutic agents. The mechanism of action by which the proliferation of the target cells occurs is essentially a mechanism of inhibition of several tyrosine kinase receptors, and especially an inhibition of the vascular endothelial growth factor receptor (VEGFR).




EXAMPLE 1Synthesis of the 6-methoxycarbonyl-2-oxindole in accordance with the process shown in synthesis scheme CSynthesis of benzoic acid, 4-chloro-3-nitro-, methylester
Synthesis of propanedioic acid, [4-(methoxycarbonyl)-2-nitrophenyl]-, dimethylester
Synthesis of 6-methoxycarbonyl-2-oxindole
A solution of 13 kg propanedioic acid, [4-(methoxycarbonyl)-2-nitrophenyl]-, dimethylester (41.77 mol) in 88 L acetic acid is hydrogenated at 45° C. and under 40-50 psi in the presence of 1.3 kg Pd/C 10%. After standstill of the hydrogenation, the reaction is heated up to 115° C. for 2 hours. The catalyst is filtered off and 180 L water is added at about 50° C. The product is isolated after cooling to 5° C., centrifugation and drying at 50° C.
EXAMPLE 2Synthesis of the “chlorimide” (methyl-1-(chloroacetyl)-2-oxoindoline-6-carboxylate)
Method 1
6-methoxycarbonyl-2-oxindole (400 g; 2.071 mol) is suspended in toluene (1200 ml) at room temperature. Chloroacetic anhydride (540 g; 3.095 mol) is added to this suspension. The mixture is heated to reflux for 3 h, then cooled to 80° C. and methyl cyclohexane (600 ml) is added within 30 min. The resulting suspension is further cooled down to room temperature within 60 min. The mother liquor is separated and the solid is washed with ice cold methanol (400 ml). The crystals are dried to afford 515.5 g (93.5%) of the “chlorimide” compound as a white solid. 1H-NMR (500 MHz, DMSO-d6) δ: 8.66 (s, 1H, 6-H); 7.86 (d, J=8.3 Hz, 1H, 8-H); 7.52 (d, J=8.3 Hz, 1H, 9-H); 4.98 (s, 2H, 15-H2); 3.95 (s, 3H, 18-H3); 3.88 (s, 2H, 3-H2). 13C-NMR (126 MHz, DMSO-d6) δ: 174.7 (C-2); 36.0 (C-3); 131.0 (C-4); 140.8 (C-5); 115.7 (C-6); 128.9 (C-7); 126.1 (C-8); 124.6 (C-9); 166.6 (C-10); 165.8 (C-13); 46.1 (C-15); 52.3 (C-18). MS: m/z 268 (M+H)+. Anal. calcd. for C12H10ClNO4: C, 53.85; H, 3.77; Cl, 13.25; N, 5.23. Found: C, 52.18; H, 3.64; Cl, 12.89; N, 5.00.
Method 2
6-Methoxycarbonyl-2-oxindole (10 g; 0.052 mol) is suspended in n-butyl acetate (25 ml) at room temperature. To this suspension a solution of chloroacetic anhydride (12.8 g; 0.037 mol) in n-butyl acetate (25 ml) is added within 3 min. The mixture is heated to reflux for 2 h, then cooled to 85° C. and methyl cyclohexane (20 ml) is added. The resulting suspension is further cooled down to room temperature and stirred for 2 h. The mother liquor is separated and the solid is washed with methanol (400 ml) at ambient temperature. The crystals are dried to afford 12.7 g (91.5%) of the “chlorimide” compound as a slightly yellow solid.
EXAMPLE 3Synthesis of the “chlorenol” (methyl-1-(chloroacetyl)-3-[methoxy(phenyl)methylene]-2-oxoindoline-6-carboxylate)
Method 1
Methyl-1-(chloroacetyl)-2-oxoindoline-6-carboxylate (12.0 g; 0.045 mol) is suspended in toluene (60 ml) at ambient temperature. Acetic anhydride (16.2 g; 0.157 mol) is added to this suspension. The mixture is heated to not less than 104° C. and trimethyl orthobenzoate (20.0 g; 0.108 mol) is added within 60 min. During the addition period and subsequent stirring at the same temperature for 3 h, volatile parts of the reaction mixture are distilled off. The concentration of the reaction mixture is kept constant by replacement of the distilled part by toluene (40 ml). The mixture is cooled down to 5° C., stirred for 1 h and filtrated. The solid is subsequently washed with toluene (14 ml) and with a mixture of toluene (8 ml) and ethyl acetate (8 ml). After drying, 16.3 g (91.7%) of the “chlorenol” compound are isolated as slightly yellow crystals. 1H-NMR (500 MHz, DMSO-d6) δ: 8.73 (d, J=1.5 Hz, 1H, 6-H); 8.09 (d, J=8.0 Hz, 1H, 9-H); 7.90 (dd, J=8.1; 1.5 Hz, 1H, 8-H); 7.61-7.48 (m, 5H, 21-H, 22-H, 23-H, 24-H, 25-H); 4.85 (s, 2H, 18-H2); 3.89 (s, 3H, 27-H3); 3.78 (s, 3H, 15-H3). 13C-NMR (126 MHz, DMSO-d6) δ: 165.9 (C-2+C16); 103.9 (C-3); 127.4; 128.6; 130.0; 135.4 (C-4+C-5+C-7+C-20); 115.1 (C-6); 126.1 (C-8); 122.5 (C-9); 166.7 (C-10); 173.4 (C-13); 58.4 (C-15); 46.4 (C-18); 128.6 (C-21+C-22+C-24+C-25); 130.5 (C-23); 52.2 (C-27). MS: m/z 386 (M+H)+. Anal. calcd. for C20H16ClNO5: C, 62.27; H, 4.18; Cl, 9.19; N, 3.63. Found: C, 62.21; H, 4.03; Cl, 8.99; N, 3.52.
Method 2
Methyl-1-(chloroacetyl)-2-oxoindoline-6-carboxylate (12.0 g; 0.045 mol) is suspended in xylene (60 ml) at ambient temperature. Acetic anhydride (16.2 g; 0.157 mol) is added to this suspension. The mixture is heated to reflux, trimethyl orthobenzoate (20.0 g; 0.108 mol) is added within 40 min and heating is maintained for 4 h. The mixture is cooled down to 0° C. and the mother liquor is separated. The solid is subsequently washed with xylene (14 ml) and a mixture of xylene (8 ml) and ethyl acetate (8 ml). After drying 14.3 g (81.0%) of the “chlorenol” compound are isolated as yellow crystals.
Method 3
Methyl-1-(chloroacetyl)-2-oxoindoline-6-carboxylate (12.0 g; 0.045 mol) is suspended in toluene (60 ml) at ambient temperature. Acetic anhydride (16.2 g; 0.157 mol) is added to this suspension. The mixture is heated to reflux, trimethyl orthobenzoate (20.0 g; 0.108 mol) is added within 40 min and heating is maintained for 3 h. The mixture is cooled down to 0° C. and the mother liquor is separated. The solid is subsequently washed with toluene (14 ml) and a mixture of toluene (8 ml) and ethyl acetate (8 ml). After drying 15.3 g (87.3%) of the “chlorenol” compound are isolated as fawn crystals.
EXAMPLE 4Synthesis of the “enolindole” (methyl-3-[methoxy(phenyl)methylene]-2-oxoindoline-6-carboxylate)
Method 1
A solution of potassium hydroxide (0.41 g, 0.006 mol) in methanol (4 ml) is added at 63° C. to a suspension of methyl-1-(chloroacetyl)-3-[methoxy(phenyl)methylene]-2-oxoindoline-6-carboxylate (8.0 g; 0.020 mol) in methanol (32 ml). The mixture is then stirred for 30 min, cooled to 0° C. and stirring is maintained for 2 h. After filtration, the solid is washed with methanol (24 ml) and dried to afford 6.0 g (94.6%) of the “enolindole” compound as yellow crystals. 1H-NMR (500 MHz, CDCl3) δ: 8.08 (s, 1H, 1-H); 7.88 (d, J=7.8 Hz, 1H, 9-H); 7.75 (m, 1H, 8-H); 7.52-7.56 (m, 3H, 18-H, 19-H, 20-H); 7.40-7.45 (m, 3H, 6-H, 17-H, 21-H); 3.92 (s, 3H, 23-H3); 3.74 (s, 3H, 13-H3). 13C-NMR (126 MHz, CDCl3) δ: 168.8 (C-2); 107.4 (C-3); 130.8 (C-4); 138.2 (C-5); 109.4 (C-6); 128.2 and 128.3 (C-7, C-16); 123.5 (C-8); 123.1 (C-9); 170.1 (C-11); 57.6 (C-13); 167.2 (C-14); 128.7 and 128.9 (C-17, C-18, C-20, C-21); 130.5 (C-19); 52.1 (C-23). MS (m/z): 310 (M+H)+. Anal. calcd. for C18H15NO4: C, 69.89; H, 4.89; N, 4.53. Found: C, 69.34; H, 4.92; N, 4.56.
Method 2
A suspension of methyl-1-(chloroacetyl)-3-[methoxy(phenyl)methylene]-2-oxoindoline-6-carboxylate (7.0 g; 0.018 mol) in methanol (28 ml) is heated to reflux. Within 3 min, a solution of sodium methoxide in methanol (0.24 g, 30 (w/w), 0.001 mol) is added to this suspension. The mixture is then stirred for 30 min, cooled to 5° C. and stirring is maintained for 2 h. After filtration, the solid is washed with methanol (9 ml) and dried to afford 5.4 g (89.7%) of the “enolindole” compound as yellow crystals.
Method 3
A suspension of methyl-1-(chloroacetyl)-3-[methoxy(phenyl)methylene]-2-oxoindoline-6-carboxylate (8.0 g; 0.021 mol) in methanol (32 ml) is heated to reflux. A solution of sodium methoxide in methanol (0.74 g, 30% (w/w), 0.004 mol), further diluted with methanol (4 ml), is added dropwise to this suspension. The mixture is then stirred for 90 min, cooled to 0° C. and stirring is maintained for 2 h. After filtration, the solid is washed with methanol (24 ml) and dried to afford 5.9 g (91.2%) of the “enolindole” compound as yellow crystals.
EXAMPLE 5Synthesis of the “chloroacetyl” (N-(4-nitroanilino)-N-methyl-2-chloro-acetamide)
Method 1
A suspension of N-methyl-4-nitroaniline (140 g; 0.920 mol) in ethyl acetate (400 ml) is heated to 70° C. Within 90 min, chloro acetylchloride (114 g; 1.009 mol) is added to this suspension. The resulting solution is then refluxed for 1 h, cooled to 60° C. and methyl cyclohexane (245 ml) is added. The suspension is further cooled down to 0° C. and stirred for 1 h. The reaction mixture is filtrated, washed with methyl cyclohexane (285 ml) and the precipitate is dried to afford 210.4 g (92.7%) of the “chloroacetyl” compound as white crystals. 1H-NMR (500 MHz, DMSO-d6) δ: 8.29 (d, J=8.5 Hz, 2H, 1-H+3-H); 7.69 (d, J=8.5 Hz, 2H, 4-H+6-H); 4.35 (s, 2H, 9-H2); 3.33 (s, 3H, 12-H3). 13C-NMR (126 MHz, DMSO-d6) δ: 124.6 (C-1+C-3); 145.6 (C-2); 127.4 (C-4+C-6); 148.6 (C-5); 165.6 (C-8); 42.7 (C-9); 37.2 (C-12). MS (m/z): 229 (M+H)+. Anal. calcd. for C9H9ClN2O3: C, 47.28; H, 3.97; N, 12.25. Found: C, 47.26; H, 3.99; Cl, 15.73; N, 12.29.
Method 2
A suspension of N-methyl-4-nitroaniline (20.0 g; 0.131 mol) in ethyl acetate (20 ml) is heated to 60° C. Within 15 min, a solution of chloro acetic anhydride (26.0 g; 0.151 mol) in ethyl acetate (60 ml) is added to this suspension. The resulting solution is then refluxed for 1 h, cooled to 75° C. ° C. and methyl cyclohexane (80 ml) is added. After seeding at 60° C., the suspension is further cooled down to 0° C. and stirred for 1 h. The reaction mixture is filtrated, washed with methyl cyclohexane (40 ml) and the precipitate is dried to afford 25.9 g (83.3%) of the “chloroacetyl” compound as grey crystals.
EXAMPLE 6Synthesis of the “nitroaniline” (N-(4-nitrophenyl)-N-methyl-2-(4-methylpiperazin-1-yl)acetamide) and of the “aniline” (N-(4-aminophenyl)-N-methyl-2-(4-methylpiperazin-1-yl)acetamide)
Method 1
A suspension of N-(4-nitroanilino)-N-methyl-2-chloro-acetamide (20.0 g; 0.087 mol) in toluene (110 ml) is heated to 40° C. Within 30 min, 1-methylpiperazine (21.9 g; 0.216 mol) is added dropwise. After purging of the dropping funnel with toluene (5 ml) the reaction mixture is stirred for 2 h at 55° C., cooled to ambient temperature and washed with water (15 ml). The organic layer is diluted with isopropanol (100 ml) and Pd/C (10%; 1.0 g) is added. After subsequent hydrogenation (H2, 4 bar) at 20° C. the catalyst is removed. Approximately ⅘ of the volume of the resulting solution is evaporated at 50° C. The remaining residue is dissolved in ethyl acetate (20 ml) and toluene (147 ml) heated to 80° C., then cooled to 55° C. and seeded. The reaction mixture is further cooled to 0° C. and stirred for 3 h at the same temperature. After filtration, the solid is washed with ice cold toluene (40 ml) and dried to afford 20.2 g (88.0%) of the “aniline” compound as white crystals. 1H-NMR (500 MHz, DMSO-d6) δ: 6.90 (d, J=8.5 Hz, 2H, 4-H+6-H); 6.65 (d, J=8.5 Hz, 2H, 1-H+3-H); 5.22 (2H, 19-H2); 3.04 (s, 3H, 9-H3); 2.79 (s, 2H, 11-H2); 2.32 (m, 4H, 13-H2+17-H2); 2.23 (m, 4H, 14-H2+16-H2); 2.10 (s, 3H, 18-H3). 13C-NMR (126 MHz, DMSO-d6) δ: 114.0 (C-1+C-3); 148.0 (C-2); 127.6 (C-4+C-6); 131.5 (C-5); 168.9 (C-8); 36.9 (C-9); 58.5 (C-11); 52.4 (C-13+C-17); 54.6 (C-14+C-16); 45.7 (C-18). MS (m/z): 263 (M+H)+. Anal. calcd. for C14H22N4O: C, 64.09; H, 8.45; N, 21.36. Found: C, 64.05; H, 8.43; N, 21.39.
Method 2
A suspension of N-(4-nitroanilino)-N-methyl-2-chloro-acetamide (14.5 g; 0.063 mol) in ethyl acetate (65 ml) is heated to 40° C. Within 30 min, 1-methylpiperazine (15.8 g; 0.156 mol) is added dropwise. After purging of the dropping funnel with ethyl acetate (7 ml) the reaction mixture is stirred at 50° C. for 90 min, cooled to ambient temperature and washed with water (7 ml). The organic layer is diluted with isopropanol (75 ml) and dried over sodium sulphate. After separation of the solid, Pd/C (10%; 2.0 g) is added and the solution is hydrogenated (H2, 5 bar) at ambient temperature without cooling. Subsequently the catalyst is removed by filtration and the solvent is evaporated at 60° C. The remaining residue is dissolved in ethyl acetate (250 ml) and recrystallized. After filtration and drying 10.4 g (60.4%) of the “aniline” compound are isolated as white crystals.
EXAMPLE 7Synthesis of the “anilino” (3-Z-[1-(4-(N-((4-methyl-piperazin-1-yl)-methylcarbonyl)-N-methyl-amino)-anilino)-1-phenyl-methylene]-6-methoxycarbonyl-2-indolinone)
Method 1
A suspension of methyl-3-[methoxy(phenyl)methylene]-2-oxoindoline-6-carboxylate (10.0 g; 0.032 mol) and N-(4-aminophenyl)-N-methyl-2-(4-methylpiperazin-1-yl)acetamide (8.6 g; 0.032 mol) in a mixture of methanol (72 ml) and N,N-dimethylformamide (18 ml) is heated to reflux. After 7 h of refluxing the suspension is cooled down to 0° C. and stirring is maintained for additional 2 h. The solid is filtered, washed with methanol (40 ml) and dried to afford 15.4 g (88.1%) of the “anilino” compound as yellow crystals. 1H-NMR (500 MHz, DMSO-d6) δ: 11.00 (s, 1H, 23-H); 12.23 (s, 19-H); 7.61 (t; J=7.1 Hz, 1H, 33-H); 7.57 (t, J=7.5 Hz, 2H, 32-H+34-H); 7.50 (d, J=7.7 Hz, 2H, 31-H+35-H); 7.43 (d, J=1.6 Hz, 1H, 29-H); 7.20 (dd, J=8.3; 1.6 Hz, 1H, 27-H); 7.13 (d, J=8.3 Hz, 2H, 14-H+18-H); 6.89 (d, 8.3 Hz, 2H, 15-H+17-H); 5.84 (d, J=8.3 Hz, 1H, 26-H); 3.77 (s, 3H, 40-H3); 3.06 (m, 3H, 12-H3); 2.70 (m, 2 H, 8-H2); 2.19 (m, 8H, 2-H2, 3-H2, 5-H2, 6-H2); 2.11 (s, 3H, 7-H3). 13C-NMR (126 MHz, DMSO-d6) δ: 54.5 (C-2+C-6); 52.2 (C-3+C-5); 45.6 (C-7); 59.1 (C-8); 168.5 (C-9); 36.6 (C-12); 140.1 (C-13); 127.6 (C-14+C-18); 123.8 (C-17+C-15); 137.0 (C-16); 158.3 (C-20); 97.5 (C-21); 170.1 (C-22); 136.2 (C-24); 128.9 (C-25); 117.2 (C-26); 121.4 (C-27); 124.0 (C-28); 109.4 (C-29); 131.9 (C-30); 128.4 (C-31+C-35); 129.4 (C-32+C-34); 130.4 (C-33); 166.3 (C-37); 51.7 (C-40). MS (m/z): 540 (M+H)+. Anal. calcd. for C31H33N5O4: C, 69.00; H, 6.16; N, 12.98. Found: C, 68.05; H, 6.21; N, 12.81.
Method 2
A suspension of methyl-3-[methoxy(phenyl)methylene]-2-oxoindoline-6-carboxylate (20.0 g; 0.064 mol) and N-(4-aminophenyl)-N-methyl-2-(4-methylpiperazin-1-yl)acetamide (17.1 g; 0.065 mol) in methanol (180 ml) is heated to reflux for 7.5 h. The resulting suspension is cooled down to 10° C. within 1 h and stirring is maintained for 1 h. After filtration, the solid is washed with ice cold methanol (80 ml) and dried to afford 31.0 g (89.0%) of the “anilino” compound as yellow crystals.
EXAMPLE 8Synthesis of the 3-Z-[1-(4-(N-((4-methyl-piperazin-1-yl)-methylcarbonyl)-N-methyl-amino)-anilino)-1-phenyl-methylene]-6-methoxycarbonyl-2-indolinone, monoethanesulfonate
A suspension of 3-Z-[1-(4-(N-((4-methyl-piperazin-1-yl)-methylcarbonyl)-N-methyl-amino)-anilino)-1-phenyl-methylene]-6-methoxycarbonyl-2-indolinone (30.0 g; 0.055 mol) in methanol (200 ml) and water (2.4 ml) is heated to 60° C. Aqueous ethanesulfonic acid (70% (w/w); 8.75 g; 0.056 mol) is added to the reaction mixture. The resulting solution is cooled to 50° C., seeded and then diluted with isopropanol (200 ml). The mixture is further cooled to 0° C. and stirred for 2 h at this temperature. The precipitate is isolated, washed with isopropanol (120 ml) and dried to furnish 35.1 g (97.3%) of the monoethanesulfonate salt of the compound as yellow crystals. 1H-NMR (400 MHz, DMSO-d6) δ: 12.26 (s, 11-H); 10.79 (s, 1H, 1-H); 9.44 (s, 1H, 24-H); 7.64 (m, 1H, 32-H); 7.59 (m, 2H, 31-H+33-H); 7.52 (m, 2H, 30-H+34-H); 7.45 (d, J=1.6 Hz, 1H, 7-H); 7.20 (dd, J=8.2; 1.6 Hz, 1H, 5-H); 7.16 (m, 2H, 14-H+16-H); 6.90 (m, 2H, 13-H+17-H); 5.85 (d, J=8.2 Hz, 1H, 4-H); 3.78 (s, 3H, 37-H3); 3.45-2.80 (broad m, 4H, 23-H2+25-H2); 3.08 (s, 3H, 28-H3); 2.88 (s, 2H, 20-H2); 2.85-2.30 (broad m, 4H, 22-H2+26-H2); 2.75 (s, 3H, 27-H3); 2.44 (q, J=7.4 Hz, 2H, 39-H2); 1.09 (t, J=7.4 Hz, 3H, 38-H3). 13C-NMR (126 MHz, DMSO-d6) δ: 9.8 (C-38); 36.6 (C-28); 42.3 (C-27); 45.1 (C-39); 51.7 (C-37); 48.9 (C-22+C-26); 52.6 (C-23+C-25); 57.5 (C-20); 97.7 (C-3); 109.5 (C-7); 117.3 (C-4); 121.4 (C-5); 123.8 (C-13+C-17); 124.1 (C-6); 127.7 (C-14+C-16); 128.4 (C-30+C-34); 128.8 (C-9); 129.5 (C-31+C-33); 130.5 (C-32); 132.0 (C-29); 168.5 (C-9); 136.3 (C-8); 137.3 (C-12); 139.5 (C-15); 158.1 (C-10); 166.3 (C-35); 168.0 (C-19); 170.1 (C-2). MS (m/z): 540 (M(base)+H)+. Anal. calcd. for C33H39N5O7S: C, 60.17; H, 6.12; N, 10.63; S, 4.87. Found: C, 60.40; H, 6.15; N, 10.70; S, 4.84.
……………………
see

|
Nintedanib
|
|
| Systematic (IUPAC) name | |
|---|---|
| Methyl (3Z)-3-{[(4-{methyl[(4-methylpiperazin-1-yl)acetyl]amino}phenyl)amino](phenyl)methylidene}-2-oxo-2,3-dihydro-1H-indole-6-carboxylate | |
| Clinical data | |
| Trade names | Vargatef, Ofev |
| AHFS/Drugs.com | Consumer Drug Information |
| Pregnancy cat. |
|
| Legal status | |
| Routes | Oral and intravenous |
| Identifiers | |
| CAS number | 656247-17-5  |
| ATC code | None |
| Chemical data | |
| Formula | C31H33N5O4 |
| Mol. mass | 539.6248 g/mol |
F. Hilberg et al. Cancer Res. 2008, 68, 4774
2. M. Reck et al. Ann. Oncol. 2011, 22, 1374
3. M. Reck et al. J. Clin. Oncol. 2013 (suppl.), Abst LBA8011
4. N. H. Hanna et al. J. Clin. Oncol. 2013, 2013 (suppl.), Abst 8034
5. J.A. Ledermann et al. J. Clin Oncol. 2011, 29, 3798
6. Glioblastoma: A. Muhac et al. J. Neurooncol. 2013, 111, 205
7. O. Bouche et al. Anticancer Res. 2011, 31, 2271
8. T. Eisen et al. J. Clin. Oncol. 2013 (suppl.), Abst. 4506

| ニンテダニブエタンスルホン酸塩 Nintedanib Ethanesulfonate C31H33N5O4.C2H6O3S : 649.76 [656247-18-6] |
| US7119093 * | Jul 21, 2003 | Oct 10, 2006 | Boehringer Ingelheim Pharma Gmbh & Co. Kg | 3-Z-[1-(4-(N-((4-Methyl-piperazin-1-yl)-methylcarbonyl)-N-methyl-amino)-anilino)-1-phenyl-methylene]-6-methoxycarbonyl-2-indolinone-monoethanesulphonate and the use thereof as a pharmaceutical composition |


Olaparib
オラパリブ
奥拉帕尼
Women suffering from advanced relapsed BRCA-mutated ovarian cancer could gain access to a new treatment option after European regulators waved through AstraZeneca’s Lynparza (olaparib).
The European Commission has approved the first-in-class PARP inhibitor for the maintenance treatment of adults with platinum-sensitive relapsed BRCA-mutated high-grade serous epithelial ovarian, fallopian tube, or primary peritoneal cancer, who are in complete response or partial response to platinum-based chemotherapy.

4-[[3-[4-(cyclopropanecarbonyl)piperazine-1-carbonyl]-4-fluorophenyl]methyl]-2H-phthalazin-1-one, cas 763113-22-0
KU-0059436
KU-59436
Olaparib (AZD-2281, trade name Lynparza) is an experimental chemotherapeutic agent, developed by KuDOS Pharmaceuticalsand later by AstraZeneca, that is currently undergoing clinical trials. It is an inhibitor of poly ADP ribose polymerase (PARP), an enzyme involved in DNA repair.[1] It acts against cancers in people with hereditary BRCA1 or BRCA2 mutations, which includes many ovarian, breast and prostate cancers.
Olaparib is an oral poly-ADP-ribose polymerase (PARP) enzyme inhibitor developed by AstraZeneca. The product is awaiting registration in the E.U. and US as a maintenance treatment of patients with BRCA mutated platinum-sensitive relapsed serous ovarian cancer. In 2014, positive opinion was received in the E.U. recommending Lynparza approval for the maintanance treatment of BRCA mutated platinum-sensitive relapsed serous ovarian cancer.
An oral poly (ADP ribose) polymerase (PARP) inhibitor being investigated by British drug company AstraZeneca, is seeking approval from the U.S. Food and Drug Administration (FDA) for the treatment of BRCA mutated platinum-sensitive relapsed ovarian cancer. AstraZeneca filed the US regulatory submission for olaparib in February 2014. Olaparib, one of several cancer drugs AstraZeneca flagged as having strong potential in its defense of a $118 billion take-over bid by Pfizer,was accepted for priority review on April 30, 2014 by the U.S. Food and Drug Administration (FDA). The NDA filing was based on Phase II study 19 data, a randomized, double-blind, placebo-controlled, Phase II study.
On June 25, 2014, FDA Oncologic Drugs Advisory Committee (ODAC), an advisory panel to the U.S. Food and Drug Administration (FDA), voted 11 to two against the accelerated approval of the PARP inhibitor olaparib as a maintenance therapy for women with platinum-sensitive relapsed ovarian cancer who have the germline BRCA (gBRCA) mutation, and who are in complete or partial response to platinum-based chemotherapy. By voting no, the committee recommended waiting for results from the larger confirmatory phase III SOLO-2 trial, which began enrolling in September 2013. According to clincialtrials.gov, the SOLO-2 study (NCT01874353) is slated to wrap in July 2015.
In terms of clinical development, phase III trials are ongoing at AstraZeneca for the treatment of gastric cancer and metastatic breast cancer. Olaparib is also in phase II clinical studies for several indications, including breast cancer, pancreatic cancer and castration-resistant prostate cancer. In March 2014, a phase II was also initiated in GB for the treatment of patients with stage IIIB or stage IV NSCLC that is not amenable to curative therapy. A phase I clinical trial for the treatment of melanoma has been completed. Phase II clinical trials are ongoing at General Hospital Corp. for the treatment of sarcoma. The drug had been in phase II clinical trials for the treatment of colorectal cancer; however no recent developments have been reported.
Discovered by KuDOS Pharmaceuticals, has experienced several twists and turns during its clinical development. Promising results for the drug were reported at the 2011 ASCO Annual Meeting, based on impressive early phase II results, only to have clinical development discontinued later that year after disappointing phase II trial results in a more generalized group of ovarian cancer patients. However, a re-analysis of the data in BRCA-positive patients – coupled with a reformulation of the drug – convinced the British drugmaker to think again and keep it going. AstraZeneca initiates Phase III clinical studies (SOLO 1 and SOLO 2) for olaparib in the U.S. in September 2013. AstraZeneca has filed Marketing Authorisation Application (MAA) for olaparib in EU in September 2013 based on Phase II study 19 data. The U.S. Food and Drug Administration has already granted olaparib orphan drug status for ovarian cancer and will hold an advisory panel hearing on the company’s application on June 25, 2014.
In 2013, orphan drug designation in the U.S. was assigned to the compound for the treatment of ovarian cancer. The compound was originally developed by Kudos Pharmaceuticals, which was acquired by AstraZeneca in 2006.
Early Phase I trials were promising, and olaparib underwent Phase II trials. However, in December 2011, AstraZeneca announced following interim analysis of a phase-II study which indicated that the previously reported progression free survival benefit was unlikely to translate into an overall survival benefit, that it would not progress into Phase III development for the maintenance treatment of serous ovarian cancer,[2] and took a charge of $285 million. The decision to discontinue development of the drug was reversed in 2013,[3] with AstraZeneca posting a new Phase III trial of Olaparib for patients with BRCA mutated ovarian cancer in April 2013.[4]
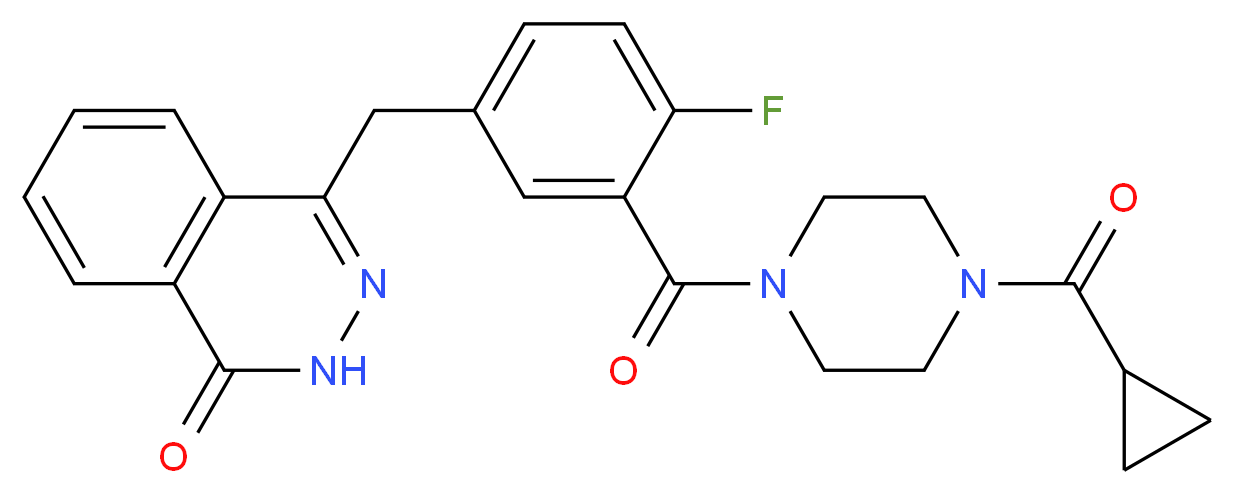
Olaparib acts as an inhibitor of the enzyme Poly ADP ribose polymerase (PARP) and is one of the first PARP inhibitors. Patients with BRCA1/2 mutations may be genetically predisposed to developing some forms of cancer, and are often resistant to other forms of cancer treatment, but this also sometimes gives their cancers a unique vulnerability, as the cancer cells have increased reliance on PARP to repair their DNA and enable them to continue dividing. This means that drugs which selectively inhibit PARP may be of significant benefit in patients whose cancers are susceptible to this treatment.[5][6][7][8][9][10]
Phase I clinical trials, in patients with BRCA-mutated tumors including ovarian cancer, were encouraging.[11] In one of these studies, it was given to 19 patients with inherited forms of advanced breast, ovarian and prostate cancers caused by mutations of the BRCA1 and BRCA2 genes. In 12 of the patients, none of whom had responded to other therapies, tumours shrank or stabilised.[12] One of the first patients to be given the treatment (who had castration-resistant prostate cancer) was as of July 2009 still in remission after two years.
In 2009 Phase II clinical trials examining the efficacy of Olaparib in treating breast, ovarian and colorectal cancer were initiated.[13][14] A phase II trial that included 63 cases of ovarian cancer concluded that olaparib is promising for women with ovarian cancer. [7 responses in 17 patients with BRCA1 or BRCA2 mutations and 11 responses in the 46 who did not have these mutations.][15]
Olaparib is generally well tolerated, the side effects consist mainly of fatigue, somnolence, nausea, loss of appetite and thrombocytopenia.

………………………

…………….
| LOU Xi-yu, YANG Xuan, DING Yi-li, WANG Jian-jun, YAN Qing-yan, HUANG Xian-gui, GUO Yang-hui, WANG Xiang-jing, XIANG Wen-sheng Synthesis of Olaparib Derivatives and Their Antitumor Activities  |
| 2013 Vol. 29 (2): 231-235 [摘要] ( 390 ) [HTML 1KB] [PDF 0KB] ( 22 ) doi: 10.1007/s40242-013-2448-5 |
……………………….

…………………
4-[3-(4-Cyclopropanecarbonylpiperazine-1-carbonyl)-4-fluorobenzyl]-2H-phthalazin-1-one: A novel bioavailable inhibitor of poly(ADP-ribose) polymerase-1
J Med Chem 2008, 51(20): 6581
…………………………..
http://www.google.co.in/patents/WO2004080976A1?cl=en
Synthesis of Key Intermediates
3- (4-0x0-3 , 4-dihydrophthalazin-l -ylmethyl) benzoic a cid (A)
A mixture of 27% sodium methoxide solution in methanol (400 g, 2 mol) and methanol (150 ml) was added dropwise between ambient temperature and 30°C over 15 minutes to a stirred mixture of phthalide (67 g, 0.5 mol), 3-formylbenzonitrile (65.5 g, 0.5 mol) and ethyl propionate (250 ml) , the mixture was stirred at ambient temperature for 40 minutes and at reflux temperature for 1 hour, then it was allowed to cool to ambient temperature. The resulting red solid was collected by filtration, washed with ethyl acetate (2 x 50 ml) and dissolved in water (1800 ml) . The solution was acidified by the addition of acetic acid (60 ml) and the resulting red solid was collected by filtration, washed with water (2 x 200 ml) and dried in vacuo to give 3- (1,3- dioxoindan-2-yl) benzonitrile (83.2 g) as a dark red solid, m.pt. 179- 182°C, m/z (M+H)+‘ 248, which was used without further purification.
3- (1, 3-Dioxoindan-2-yl) benzonitrile (74.18 g, 0.3 mol) was added in portions to a solution of sodium hydroxide (36 g, 0.9 mol) in water (580 ml), the resulting dark red suspension was stirred at reflux temperature for 5 hours, then it was cooled to ambient temperature and washed with ethyl acetate (3 x 300 ml) . The aqueous solution was acidified by the dropwise addition of concentrated hydrochloric acid (110 ml), the mixture was stirred at ambient temperature for 1 hour, then the resulting solid was collected by filtration, washed with water (2 x 200 ml) and dried in vacuo to give a 1:1 mixture of 3- (1,3- dioxoindan-2-yl)benzoic acid, (M+H)+” 267, and 2- [2- (3- carboxyphenyl) acetyl] benzoic acid, (M+H)+‘ 285, (69.32 g) , which was used without further purification.
The mixture obtained in the previous step (52.8 g) was added to a solution of triethylamine (37.55 g, 0.372 mol) in industrial methylated spirit (500 ml) and the resulting cloudy solution was filtered through a pad of filter-aid to give a clear solution. Hydrazine monohydrate (9.3 g, 0.186 mol) was added in one portion at ambient temperature, the stirred mixture was heated under reflux for 1 hour, then it was concentrated in vacuo to approximately 250 ml and added to a solution of sodium acetate (41 g, 0.5 mol) in water (500 ml) . The mixture was brought to pH 7 by the dropwise addition of concentrated hydrochloric acid, then it was stirred at ambient temperature for 3 hours. The resulting solid was collected by filtration, washed with water (50 ml) and dried in va cuo to give a white solid (15.62 g) . The combined filtrate and washings were acidified to pH 6 by the addition of hydrochloric acid, then the mixture was stirred at ambient temperature for 3 hours. The resulting solid was collected by filtration, washed with water (50 ml) and dried in va cuo to give a second crop of off-white solid (17.57 g) . The combined filtrate and washings from the second crop were readjusted to pH 6 and treated as before to give a third crop of pale orange solid (6.66 g) . The three crops were combined to give essentially pure 3- (4-oxo-3, 4-dihydrophthalazin-l-ylmethyl) benzoic acid (A), (M+H)+‘ 281, δH 4.4 (2H, s), 7.2-7.4 (IH, m) , 7.5-7.6 (IH, ) , 7.7-8.0 (5H, m) , 8.1- 8.2 (IH, m) , 12.6 (IH, s)
b . 2-Fluoro-5- (4-oxo-3 , 4-dihydro-phthalazin -l -ylmethyl) benzoi c a cid (B)
Dimethyl phosphite (22.0 g, 0.2 mol) was added drop-wise to a solution of sodium methoxide (43.0 g) in methanol (100 ml) at 0°C. 2- Carboxybenzaldehyde (21.0 g, 0.1 mol) was then added portion-wise to the reaction mixture as a slurry in methanol (40 ml), with the temperature kept below 5°C. The resulting pale yellow solution was warmed to 20°C over 1 hour. Methanesulphonic acid (21.2 g, 0.22 mol) was added to the reaction drop-wise and the resulting white suspension was evaporated in va cuo . The white residue was quenched with water and extracted into chloroform (3 x 100 ml) . The combined organic extracts were washed with water (2 x 100 ml) , dried over MgS04, and evaporated in va cuo to yield (3-oxo-l, 3-dihydro-isobenzofuran-l-yl) phosphonic acid dimethyl ester as a white solid (32.0 g, 95 %, 95 % purity) . This was then used without further purification in the next stage.
To a mixture of (3-oxo-l, 3-dihydro-isobenzofuran-l-yl) phosphonic acid dimethyl ester (35.0 g, 0.14 mol) in tetrahydrofuran (200 ml) and 2- fluoro-5-formylbenzonitrile (20.9 g, 0.14 mol) in tetrahydrofuran (130 ml) was added triethylamine (14 ml, 0.14 mol) drop-wise over 25 min, with the temperature kept below 15°C. The reaction mixture was warmed slowly to 20°C over 1 hour and concentrated in vacuo . The white residue was slurried in water (250 ml) for 30 minutes, filtered, washed with water, hexane and ether, and dried to yield 2-fluoro-5- (3- oxo-3H-isobenzofuran-l-ylidenemethyl) benzonitrile as a 50:50 mixture of E and Z isomers (37.2 g, 96 %); m/z [M+l]+ 266 (98 % purity) To a suspension of 2-fluoro-5- (3-oxo-3H-isobenzofuran-l- ylidenemethyl) benzonitrile in water (200 ml) was added aqueous sodium hydroxide (26.1 g in 50 ml water) solution and the reaction mixture was heated under nitrogen to 90 °C for 30 minutes. The reaction mixture was partially cooled to 70°C, and hydrazine hydrate (100 ml) was added and stirred for 18 hours at 70°C. The reaction was cooled to room temperature and acidified with 2M HC1 to pH 4. The mixture was stirred for 10 min and filtered. The resulting solid was washed with water, hexane, ether, ethyl acetate and dried to yield 2-fluoro-5- (4-oxo-3, 4- dihydrophthalazin-l-ylmethyl)benzoic acid as a pale pink powder (30.0 g, 77 %) . m/z [M+l]+ 299 (96 % purity), δH 4.4 (2H, s) , 7.2-7.3 (IH, m) , 7.5-7.6 (IH, m) , 7.8-8.0 (4H, m) , 8.2-8.3 (IH, m) , 12.6 (IH, s).
c . 1 – [3- (4-Oxo-S , 4-dihydrophthalazin-l -ylmethyl) benzoyl]piperidine-4- carboxylic a cid (C)
(A) (C)
3- (4-Oxo-3, 4-dihydrophthalazin-l-ylmethyl)benzoic acid (A) (7.0 g, 0.25 mol), ethyl isonipecotate (5 ml, 0.32 mol), 2- (lH-benzotriazol-1-yl) – 1, 1, 3, 3-tetramethyluronium hexafluorophosphate (HBTU) (12.3 g, 0.32 mol) and N, N, -diisopropylethylamine (10.0 ml, 0.55 mol) were added to dimethylacetamide (40 ml) and stirred for 18 h. Water (100 ml) was added to the reaction mixture and the product was extracted into dichloromethane (4 x 50 ml) . The combined organic layers were washed with water (3 x 100 ml), dried over MgS0, filtered and evaporated in va cuo to yield an oil. To a solution of the oil in tetrahydrofuran (100 ml) was added 10 % aqueous sodium hydroxide solution (20 ml) and the reaction was stirred for 18 hours. The reaction was concentrated, washed with ethyl acetate (2 x 30 ml) and acidified with 2M HCl to pH 2. The aqueous layer was extracted with dichloromethane (2 x 100 ml), then the extracts were dried over MgS04, filtered and evaporated to yield 1- [3- (4-oxo-3, 4-dihydrophthalazin-l-ylmethyl)benzoyl]piperidine- 4-carboxylic acid (C) as a yellow solid (7.0 g, 65 %), m/z [M+l]+ 392
(96 % purity), δH 1.3-1.8 (5H, m) , 2.8-3.1 (4H, m) , .4 (2H, s), 7.2- 7.3 (IH, m) , 7.3-7.4 (IH, ) , 7.7-8.0 (5H, m) , 8.2-E 3 (IH, m) , 12.6 (IH, s) .
d . 1 – [2-Fluoro-5- (4 -oxo-3 , 4-dihydrophthala zin-l – ylmethyl) benzoyl]piperidine-4~carboxylic a cid (D)
(B) (D)
2-Fluoro-5- ( -oxo-3, 4-dihydrophthalazin-l-ylmethyl) benzoic acid (B) (3.1 g, 0.14 mol), ethyl isonipecotate (1.7 ml, 0.11 mol), 2-(lH- benzotriazol-1-yl) -1,1,3, 3-tetramethyluronium hexafluorophosphate (HBTU) (5.1 g, 0.13 mol) and N,N, -diisopropylethylamine (10.0 ml, 0.55 mol) were added to dimethylacetamide (15 ml) and stirred for 18 hours. Water (100 ml) was added to the reaction mixture and the product was extracted into dichloromethane (4 x 50 ml) . The combined organic layers were, filtered, washed with water (3 x 100 ml), dried over MgS04, filtered and evaporated in vacuo to yield an orange oil. The oil was purified by flash chromatography (ethyl acetate) to yield l-[2- fluoro-5- (4-oxo-3, 4-dihydrophthalazin-l-ylmethyl) benzoyl] piperidine-4- carboxylic acid as the methyl ester (1.5 g, 33 %, 96 % purity) . To a solution of the methyl ester in tetrahydrofuran: water (2:1, 40 ml) was added sodium hydroxide (0.3 g, 0.075 mol) and the reaction was stirred for 18 h. The reaction was concentrated, washed with ethyl acetate (2 x 20 ml) and acidified with 2M HC1 to pH 2. The aqueous layer was extracted with dichloromethane (2 x 20 ml) , and the combined extracts were dried over MgS04 and evaporated to yield 1- [3- ( 4-oxo-3, 4- dihydrophthalazin-1-ylmethyl) benzoyl] piperidine- -carboxylic acid (D) as a yellow solid (0.6 g, 65 %), m/z [M+l]+ 392 (96 % purity) Example 1 – Synthesis of Key Compounds
a. Synthesis of 4- [3- (piperazine-1-carfoonyl)benzyl] -2H-phthalasin-l- one (1)
(A) (1)
3- (4-0xo-3, 4-dihydrophthalazin-l-ylmethyl) benzoic acid (A) (5.0g, 0.17mol), tert-butyl 1-piperazinecarboxylate (3.9 g, 0.21 mol), 2-(lH- benzotriazol-1-yl) -1,1,3, 3-tetramethyluronium hexafluorophosphate (HBTU) (8.6 g, 0.22 mol) and N, , -diisopropylethylamine (6.7 ml, 0.38 mol) were added to dimethylacetamide (40 ml) and stirred for 18 hours. Water (100 ml) was added and the reaction mixture was heated to 100°C for 1 hour. The suspension was cooled to room temperature, filtered and dried to yield a white solid. The solid was dissolved in a solution of 6M HC1 and ethanol (2:1, 50 ml) and stirred for 1 hour. The reaction was concentrated, basified with ammonia to pH 9, and the product was extracted into dichloromethane (2 x 50 ml). The combined organic layers were washed with water (2 x 50 ml), dried over MgS04, and evaporated in va cuo to yield 4- [3- (piperazine-1-carbonyl) benzyl] – 2H-phthalazin-l-one (1) as a yellow crystalline solid (4.0 g, 77 %); m/z [M+l]+ 349 (97 % purity), δH 2.6-3.8 (8H, ) , 4.4 (2H, s), 7.2-7.5 (4H, m) , 7.7-8.0 (3H, m) , 8.2-8.3 (IH, m) , 12.6 (IH, s)
b . Synthesis of 4 – [4-Fluoro-3- (piperazine-1 -carbonyl) benzyl ] -2H- phthala zin ~l -one (2)
(β) (2)
The synthesis was carried out according to the method described in (a) above using 2-fluoro-5- (4-oxo-3, -dihydrophthalazin-l-ylmethyl) benzoic acid (B) to yield 4- [4-fluoro-3- (piperazine-1-carbonyl) benzyl] -2H- phthalazin-1-one (2) as a white crystalline solid (4.8 g, 76 %); m/z [M+l]+ 367 (97 % purity), δH 2.6-3.8 (8H, m) , 4.4 (2H, s), 7.2-7.5 (3H, m) , 7.7-8.0 (3H, m) , 8.2-8.3 (IH, m) , 12.6 (IH, s) .
…………………………..
US 8183369
http://www.google.co.in/patents/US8183369
4-[3-(4-Cyclopropanecarbonyl-piperazine-1-carbonyl)-4-fluoro-benzyl]-2H-phthalazin-1-one (compound A) disclosed in WO 2004/080976:
is of particular interest.
A crystalline form of compound A (Form A) is disclosed in co-pending applications, which claim priority from U.S. 60/829,694, filed 17 Oct. 2006, entitled “Phthalazinone Derivative”, including U.S. Ser. No. 11/873,671 and WO 2008/047082.
Form A
2-Fluoro-5-[(4-oxo-3,4-dihydrophthalazin-1-yl)methyl]benzoic acid (D)(15.23 g, 51.07 mmol) was suspended with stirring under nitrogen in acetonitrile (96 ml). Diisopropylethylamine (19.6 ml, 112.3 mmol) was added followed by 1-cyclopropylcarbonylpiperazine (I)(9.45 g, 61.28 mmol) and acetonitrile (1 ml). The reaction mixture was cooled to 18° C. 0-Benzotriazol-1-yl-tetramethyluronium hexafluorophosphate (25.18 g, 66.39 mmol) was added over 30 minutes and the reaction mixture was stirred for 2 hours at room temperature. The reaction mixture was cooled to 3° C. and maintained at this temperature for 1 hour, before being filtered. The filter cake was washed with cold (3° C.) acetonitrile (20 ml) before being dried in vacuo at up to 40° C. to give the title compound as a pale yellow solid (20.21 g).
Mass Spectrum: MH+ 435
1H NMR (400 MHz, DMSO-d6) δ: 0.70 (m, 4H), 1.88 (br s, 1H), 3.20 (br s, 2H), 3.56 (m, 6H), 4.31 (s, 2H), 7.17 (t, 1H), 7.34 (dd, 1H), 7.41 (m, 1H), 7.77 (dt, 1H), 7.83 (dt, 1H), 7.92 (d, 1H), 8.25 (dd, 1H), 12.53 (s, 1H).
………………………..
http://www.google.co.in/patents/US8247416
4-[3-(4-Cyclopropanecarbonyl-piperazine-1-carbonyl)-4-fluoro-benzyl]-2H-phthalazin-1-one (compound A) disclosed in WO 2004/080976:
is of particular interest.
In WO 2004/080976, compound A was synthesised as one of a number of library compounds from 4-[4-fluoro-3-(piperazine-1-carbonyl)-benzyl]-2H-phthalazin-1-one (compound B):
by the addition of cyclopropanecarbonyl chloride:
to a solution of (B) in dichloromethane, followed by Hünig’s base (N,N-diisopropylethyl amine). This reaction is carried out with stirring at room temperature for 16 hours, and the resulting compound being purified by preparative HPLC.
The piperazine derivative (B) was prepared by deprotecting 4-[2-fluoro-5-(4-oxo-3,4-dihydro-phthalazin-1-ylmethyl)-benzoyl]-piperazine-1-carboxylic acid tert-butyl ester (compound C):
by the use of 6M HCl and ethanol for 1 hour, followed by basification with ammonia to pH 9, and extraction into dichloromethane.
The Boc-protected piperazine derivative (C) was prepared from 2-fluoro-5-(4-oxo-3,4-dihydro-phthalazin-1-ylmethyl)-benzoic acid (compound D):
by the addition of piperazine-1-carboxylic acid tert-butyl ester:
2-(1H-benzotriazol-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate (HBTU) and N,N,-diisopropylethylamine in dimethylacetamide, followed by stirring for 18 hours.
In WO 2004/080976, the following route to compound D is disclosed:
The method of synthesising compound D may further comprise the step of:
(c) synthesising 2-fluoro-5-[(4-oxo-3,4-dihydrophthalazin-1-yl)methyl]benzonitrile (ED):
from compound E by reaction with hydrazine hydrate; and
(d) synthesising compound D from compound ED by reaction with sodium hydroxide.
Step (c) may be achieved by using between 1.1 and 1.3 equivalents of hydrazine hydrate in tetrahydrofuran followed by neutralisation of the excess hydrazine hydrate using acetic acid.
A sixth aspect of the present invention provides the compound ED:
and its use in the synthesis of compound D.
EXAMPLES
Example 1Synthesis of Compound A
Starting material (D) was synthesised by the method disclosed in WO 2004/080976
Methods
Preparative HPLC
Samples were purified with a Waters mass-directed purification system utilising a Waters 600 LC pump, Waters Xterra C18 column (5 μm 19 mm×50 mm) and Micromass ZQ mass spectrometer, operating in positive ion electrospray ionisation mode. Mobile phases A (0.1% formic acid in water) and B (0.1% formic acid in acetonitrile) were used in a gradient; 5% B to 100% over 7 min, held for 3 min, at a flow rate of 20 ml/min.
Analytical HPLC-MS
Analytical HPLC was carried out with a Spectra System P4000 pump and Jones Genesis C18 column (4 μm, 50 mm×4.6 mm). Mobile phases A (0.1% formic acid in water) and B (acetonitrile) were used in a gradient of 5% B for 1 min rising to 98% B after 5 min, held for 3 min at a flow rate of 2 ml/min. Detection was by a TSP UV 6000LP detector at 254 nm UV and range 210-600 nm PDA. The Mass spectrometer was a Finnigan LCQ operating in positive ion electrospray mode.
(a) 4-[2-Fluoro-5-(4-oxo-3,4-dihydro-phthalazin-1-ylmethyl)-benzoyl]-piperazine-1-carboxylic acid tert-butyl ester (C)
To a stirred solution of the starting material D (850 g) in dimethylacetamide (DMA) (3561 ml) at room temperature under nitrogen was added HBTU (2-(1H-benzotriazol-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate) (1402 g) in one portion. Hünig’s base (iPr2NEt, 1096 ml) was then added with the temperature kept between 15 to 25° C. followed by a solution of 1-Boc-piperazine (637 g) in DMA (1428 ml) with the temperature kept between 15 to 25° C.
The solution was stirred at room temperature for 2 hours and sampled for completion (HPLC). Upon completion the solution was added to vigorously stirred water (17085 ml) with the temperature kept between 15 to 25° C. and the solid filtered off, washing with water (2×7131 ml), hexane (2×7131 ml) and methyl tert-butyl ether (MTBE) (2×3561 ml). The solid was then dried overnight and then sampled for water content and chemical purity.
This reaction was then repeated, see table:
| Purity | Water Content | |||
| Batch | Yield (g) | (HPLC Area %) | (K.F.) | Corrected yield |
| 1 | 1571.3 | 86.80 | 24.3 | 1032.5 g (78%) |
| 2 | 2781.6 | 85.00 | 40.3 | 1411.5 g (106%) |
| a. Greater than 100% yield attributed to non-representative sampling | ||||
(b) 4-[4-Fluoro-3-(piperazine-1-carbonyl)-benzyl]-2H-phthalazin-1-one (B)
To a stirred solution of industrial methylated spirits (IMS) (2200 ml) and concentrated HCl (4400 ml) was added compound C (2780.2 g) in portions at room temperature under nitrogen, the foaming was controlled by the addition rate. The solution was then stirred at 15 to 25° C. for 30 minutes and sampled for completion (HPLC).
Upon completion the solution was evaporated to remove any IMS and the aqueous extracted with CH2Cl2 (2×3500 ml) before the pH was adjusted to >8 using concentrated ammonia. The resultant slurry was then diluted with water (10000 ml) and extracted with CH2Cl2 (4×3500 ml), washed with water (2×2000 ml), dried over MgSO4 (250 g) and evaporated. The crude product was then slurried in CH2Cl2 (3500 ml) and added to MTBE (5000 ml). The resultant suspension was filtered and dried at 50° C. overnight yielding 611.0 g (58.5% yield) of material with a purity of 94.12%
(c) 4-[3-(4-Cyclopropanecarbonyl-piperazine-1-carbonyl)-4-fluoro-benzyl]-2H-phthalazin-1-one (A)
To a stirred suspension of compound B (1290 g) in CH2Cl2 (15480 ml) under nitrogen was added a pre-mixed solution of triethylamine (470 ml) and cyclopropane carbonyl chloride (306 ml) in CH2Cl2 (1290 ml) dropwise with the temperature kept below 20° C. The solution was then stirred at 10-15° C. for 15 minutes and sampled for completion. The reaction mixture was found to contain only 1.18% of starting material B and so the reaction was deemed complete and the batch was then worked-up.
The reaction mixture was washed with water (7595 ml), 5% citric acid solution (7595 ml), 5% sodium carbonate solution (7595 ml) and water (7595 ml). The organic layer was then dried over magnesium sulfate (500 g).
The CH2Cl2 containing product layer was then isolated, filtered through Celite and charged to a 251 vessel. CH2Cl2 (8445 ml) was then distilled out at atmospheric pressure and ethanol (10000 ml) added. Distillation was then continued with every 4000 ml of distillate that was removed being replaced with ethanol (4000 ml) until the head temperature reached 73.7° C. The reaction volume was then reduced (to 7730 ml) by which time the head temperature had reached 78.9° C. and the solution was allowed to cool to 8° C. overnight. The solid was then filtered off, washed with ethanol (1290 ml) and dried at 70° C. overnight. Yield=1377.3 g (90%). HPLC purity (99.34% [area %]). Contained 4.93% ethanol and 0.45% CH2Cl2 by GC.
(d) Water Treatment of Compound A
A suspension of compound A (1377.0 g), as produced by the method of Example 1, in water (13770 ml) was heated to reflux for 4 hours, cooled to room temperature and filtered. The solid was washed with water (2754 ml) and dried at 70° C. overnight. Yield=1274.8 g (92.6%). HPLC purity (99.49% [area %]). Contained 0.01% ethanol and 0.01% CH2Cl2 by GC.
1H NMR spectrum of compound A (DMSO-d6) following the water treatment is shown in FIG. 1.

The powder XRD pattern of Compound A following the water treatment is shown in FIG. 2, which shows the compound is as Form A.
Example 2
Alternative Synthesis of Compound A Using 1-(cyclopropylcarbonyl) piperazine
Methods (also for Examples 3 & 4)
NMR
1H NMR spectra were recorded using Bruker DPX 400 spectrometer at 400 MHz. Chemical shifts were reported in parts per million (ppm) on the δ scale relative to tetramethylsilane internal standard. Unless stated otherwise all samples were dissolved in DMSO-d6.
Mass Spectra
Mass spectra were recorded on an Agilent XCT ion trap mass spectrometer using tandem mass spectrometry (MS/MS) for structural confirmation. The instrument was operated in a positive ion elctrospray mode.
(a) 4-[3-(4-Cyclopropanecarbonyl-piperazine-1-carbonyl)-4-fluoro-benzyl]-2H-phthalazin-1-one (Compound A)
2-Fluoro-5-[(4-oxo-3,4-dihydrophthalazin-1-yl)methyl]benzoic acid (D)(15.23 g, 51.07 mmol) was suspended with stirring under nitrogen in acetonitrile (96 ml). Diisopropylethylamine (19.6 ml, 112.3 mmol) was added followed by 1-cyclopropylcarbonylpiperazine (1)(9.45 g, 61.28 mmol) and acetonitrile (1 ml). The reaction mixture was cooled to 18° C. O-Benzotriazol-1-yl-tetramethyluronium hexafluorophosphate (25.18 g, 66.39 mmol) was added over 30 minutes and the reaction mixture was stirred for 2 hours at room temperature. The reaction mixture was cooled to 3° C. and maintained at this temperature for 1 hour, before being filtered. The filter cake was washed with cold (3° C.) acetonitrile (20 ml) before being dried in vacuo at up to 40° C. to give the title compound as a pale yellow solid (20.21 g).
Mass Spectrum: MH+435
1H NMR (400 MHz. DMSO-d6) δ: 0.70 (m, 4H), 1.88 (br s, 1H), 3.20 (br s, 2H), 3.56 (m, 6H), 4.31 (s, 2H), 7.17 (t, 1H), 7.34 (dd, 1H), 7.41 (m, 1H), 7.77 (dt, 1H), 7.83 (dt, 1H), 7.92 (d, 1H), 8.25 (dd, 1H), 12.53 (s, 1H).
Example 3Alternative Synthesis of Compound A Using 1-(cyclopropylcarbonyl) piperazine HCl salt
(a) 1-(Cyclopropylcarbonyl)piperazine HCl salt (I′)
Acetic acid (700 ml) was treated with piperazine (50.00 g, 0.581 mol) portionwise over 15 minutes with stirring under nitrogen The reaction mixture was warmed to 40° C. and maintained at this temperature until a complete solution was obtained. Cyclopropanecarbonyl chloride 59.2 ml, 0.638 mol) was added over 15 minutes. The reaction mixture was stirred at room temperature overnight. The reaction mixture was filtered and the filtrate distilled under reduced pressure until ˜430 ml of distillates had been collected. Toluene (550 ml) was charged to the reaction mixture and reduced pressure distillation continued until a further 400 ml of distillates were collected. A further charge of toluene (550 ml) was added and reduced pressure distillation continued until 350 ml of distillates were collected. The resulting slurry was diluted with toluene (200 ml) and stirred overnight. Further toluene (500 ml) was added in order to mobilise the slurry. The slurry was filtered, washed with toluene (100 ml) and dried in vacuo at 40° C. to give the title compound as an off white solid (86.78 g).
Mass Spectrum: MH+155
1H NMR (400 MHz. D2O) δ: 0.92 (m, 4H), 1.98 (m, 1H), 3.29 (m, 2H), 3.38 (m, 2H), 3.84 (m, 2H), 4.08 (m, 2H).
(b) Compound A
2-Fluoro-5-[(4-oxo-3,4-dihydrophthalazin-1-yl)methyl]benzoic acid (D)(0.95 g, 3.19 mmol) was suspended with stirring under nitrogen in acetonitrile (4 ml). 2-(1H-benzotriazol-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate (HBTU) (1.45 g, 3.83 mmol) was added followed by 1-cyclopropylcarbonylpiperazine HCl salt (I′)(0.73 g, 3.83 mmol). Diisopropylethylamine (1.39 ml, 7.98 mmol) was added over 3 minutes and the reaction mixture was stirred for overnight at room temperature. The reaction mixture was cooled to 5° C. and maintained at this temperature for 1 hour, before being filtered. The filter cake was washed with cold (3° C.) acetonitrile (2 ml) before being dried in vacuo at up to 40° C. to give the title compound as a pale yellow solid (0.93 g).
| Patent | Submitted | Granted |
|---|---|---|
| Phthalazinone Derivatives [US2012010204] | 2012-01-12 | |
| PARP1 TARGETED THERAPY [US2012035244] | 2012-02-09 | |
| Phthalazinone derivatives [US7449464] | 2005-03-17 | 2008-11-11 |
| 4- [3- (4-CYCLOPROPANECARBONYL-PIPERAZINE-I-CARBONYL) -4 -FLUORO-BENZYL] -2H-PHTHALAZ IN-1-ONE [US8183369] | 2010-11-11 | 2012-05-22 |
| PHTHALAZINONE DERIVATIVES [US7692006] | 2008-06-19 | 2010-04-06 |
| PHTHALAZINONE DERIVATIVES [US7981889] | 2008-08-21 | 2011-07-19 |
| PHARMACEUTICAL FORMULATION 514 [US2010098763] | 2010-04-22 | |
| PHTHALAZINONE DERIVATIVE [US8247416] | 2009-10-29 | 2012-08-21 |
| WO2002036576A1 * | 25 Oct 2001 | 10 May 2002 | Kudos Pharm Ltd | Phthalazinone derivatives |
| WO2002090334A1 * | 30 Apr 2002 | 14 Nov 2002 | Kudos Pharm Ltd | Isoquinolinone derivatives as parp inhibitors |
| WO2003093261A1 * | 29 Apr 2003 | 13 Nov 2003 | Kudos Pharm Ltd | Phthalazinone derivatives |
extras…………..
|
|
| Systematic (IUPAC) name | |
|---|---|
| 4-[(3-[(4-cyclopropylcarbonyl)piperazin-4-yl]carbonyl) -4-fluorophenyl]methyl(2H)phthalazin-1-one | |
| Clinical data | |
| Trade names | Lynparza |
| Legal status |
|
| Routes | Oral |
| Identifiers | |
| CAS number | 763113-22-0 |
| ATC code | None |
| PubChem | CID 23725625 |
| ChemSpider | 23343272  |
| UNII | WOH1JD9AR8 |
| ChEMBL | CHEMBL521686 |
| Chemical data | |
| Formula | C24H23FN4O3 |
| Mol. mass | 435.08 g/mol |
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
 CAS NO. 763113-22-0, olaparib H-NMR spectral analysis |
 CAS NO. 763113-22-0, olaparib C-NMR spectral analysis |

Bafetinib
4-[[(3S)-3-(dimethylamino)pyrrolidin-1-yl]methyl]-N-[4-methyl-3-[(4-pyrimidin-5-ylpyrimidin-2-yl)amino]phenyl]-3-(trifluoromethyl)benzamide, cas 859212-16-1
4-[(S)-3-(dimethylamino)pyrrolidin-1-ylmethyl]-3-trifluoromethyl-N-{4-methyl-3-[4-(5-pyrimidinyl)pyrimidin-2-ylamino]phenyl}benzamide
859212-07-0 (hydrochloride)
Bafetinib , previously as INNO-406 , NS-187 and CNS-9 refers is an experimental drug from the substance group ofbenzamides , who as Tyrosinkinasehemmstoff to be used. [2] It was originally developed by the Japanese company Nippon Shinyaku and 2006 Innovive Pharmaceuticals licensed. [3] Innovive was established in June 2008 by the CytRx Corp. adopted. [4]
Bafetinib, also known as INNO-406, is an orally bioavailable 2-phenylaminopyrimidine derivative with potential antineoplastic activity. Bafetinib specifically binds to and inhibits the Bcr/Abl fusion protein tyrosine kinase, an abnormal enzyme produced by Philadelphia chromosomal translocation associated with chronic myeloid leukemia (CML). This agent also inhibits the Src-family member Lyn tyrosine kinase, upregulated in imatinib-resistant CML cells and in a variety of solid cancer cell types. The inhibitory effect of bafetinib on these specific tyrosine kinases may decrease cellular proliferation and induce apoptosis in tumor cells that overexpress these kinases. CML patients may be refractory to imatinib, which sometimes results from point mutations occurring in the kinase domain of the Bcr/Abl fusion product. Due to its dual inhibitory activity, the use of bafetinib has been shown to overcome this particular drug resistance.
INNO-406 (formerly NS-187) is a potent, orally available, rationally designed, dual Bcr-Abl and Lyn kinase inhibitor that is currently in early clinical studies at CytRx Oncology for the treatment of B-cell chronic lymphocytic leukemia, metastatic prostate cancer and glioblastoma multiforme. CytRx is also conducting phase I clinical studies for the treatment of recurrent high-grade glioma or metastatic disease to the brain that has progressed after treatment with whole brain radiation therapy or stereotactic radiosurgery.
The company is developing INNO-406 in preclinical studies for the prevention of bone loss in multiple myeloma patients. Nippon Shinyaku is also evaluating the compound for the treatment of chronic myeloid leukemia. The compound had been under evaluation for the treatment of certain forms of acute myeloid leukemia (AML) that are refractory or intolerant of other approved treatments; however, no recent development has been reported for this indication.
Based on its mechanisms of action, INNO-406 is expected to be effective in treating Gleevec-resistant CML and may delay or even prevent the onset of resistance in treatment naive CML patients. The ability of INNO-406 to specifically target the Bcr-Abl and Lyn kinases may result in a better side effect profile than compounds that target multiple kinases such as a pan-Src inhibitor.
In 2005, the compound was licensed to Innovive Pharmaceuticals (acquired by CytRx Oncology in 2008) by Nippon Shinyaku on a worldwide basis, with the exception of Japan, for the treatment of CML. Orphan drug designation was assigned to the compound for the treatment of CML in the U.S in 2007 and in the E.U. in 2010.
Bafetinib is an inhibitor of tyrosine kinases . It affects the formation of the fusion protein Bcr-Abl , as well as that of theenzyme Lyn kinase and should in mice ten times stronger effect than the imported Tyrosinkinasehemmstoff imatinib .[5]
| Patent | Submitted | Granted |
|---|---|---|
| Amide Derivative and Medicine [US7728131] | 2008-11-27 | 2010-06-01 |
Bafetinib currently has no indication for an authorization as medicines .
The drug is intended for the treatment of chronic lymphocytic leukemia are developed (CLL). For this indication is Bafetinib is in the development phase II (June 2011). [6]
Bafetinib is also in phase II for the treatment of hormone-refractory prostate cancer . [7]
The US regulatory authority FDA had Bafetinib end of 2006, the status of a drug orphan (orphan drug) awarded. [8]This status could allow an accelerated development and approval.

N-[3-([5,5′-Bipyrimidin]-2-ylamino)-4-methylphenyl]-4-[[(3S)-3-(dimethyl-amino)-1-pyrrolidinyl]methyl]-3-(trifluoromethyl)benzamide
CAS No .: 887650-05-7
MW: 576.62
Formula: C 30 H 31 F 3 N 8 O
Synonym: INNO-406, NS-187
4-(bromomethyl)-3-trifluoromethylbenzoic acidTo 60.0 g of 4-methyl-3-trifluoromethylbenzoic acid was added 600 ml of isopropyl acetate. Under stirring at room temperature, a solution of 133.0 g of sodium bromate in 420 ml of water and a solution of 91.7 g of sodium hydrogensulfite in 180 ml of water were added in turn. The mixture was gradually heated from 30° C. up to 50° C. at intervals of 10° C. and stirred until the color of the reaction solution disappeared. The aqueous layer was separated to remove, and to the organic layer were added a solution of 133.0 g of sodium bromate in 420 ml of water and a solution of 91.7 g of sodium hydrogensulfite in 180 ml of water, and then the mixture was gradually heated up to 60° C. as above. After separation, to the organic layer were further added a solution of 133.0 g of sodium bromate in 420 ml of water and a solution of 91.7 g of sodium hydrogensulfite in 180 ml of water, and the mixture was gradually heated as above and heated to the temperature the mixture was finally refluxed. After the completion of the reaction, the reaction solution was separated, the organic layer was washed twice with a 5% aqueous sodium thiosulfate solution and twice with 15% saline, dried over anhydrous magnesium sulfate, and, then the solvent was distilled off under reduced pressure. To the residue was added 120 ml of n-heptane, the mixture was stirred, and then the crystals were collected by filtration to obtain 50.0 g of the objective compound as colorless crystals.
Melting point: 140-143° C.
Step 2
4-(bromomethyl)-3-trifluoromethyl-N-{4-methyl-3-[4-(5-pyrimidinyl)pyrimidin-2-ylamino]phenyl}benzamide7.69 g of 4-(bromomethyl)-3-trifluoromethylbenzoic acid obtained in the step 1 was suspended in 154 ml of anhydrous dichloromethane. Under ice-cool stirring, 6.59 ml of oxalyl chloride and 0.1 ml of anhydrous N,N-dimethylformamide were added dropwise. Under ice cooling, the mixture was further stirred for 3 hours, and then the reaction solution was concentrated under reduced pressure. To the residue was added 70 ml of anhydrous 1,4-dioxane, and then 7.00 g of 4-methyl-3-[4-(5-pyrimidinyl)pyrimidin-2-ylamino]aniline (Reference Example 18) and 4.18 g of potassium carbonate were added in turn, followed by stirring at room temperature for 18 hours. To the reaction solution was added 175 ml of water, and the mixture was violently stirred for one hour. Then, the deposit was collected by filtration and washed in turn with water, a small amount of acetonitrile, ethyl acetate and diisopropyl ether to obtain 8.10 g of the objective compound as pale yellow crystals.
Melting point: 198-202° C. (with decomposition)
To a solution of 6.00 g of 4-(bromomethyl)-3-trifluoromethyl-N-{4-methyl-3-[4-(5-pyrimidinyl)pyrimidin-2-ylamino]phenyl}benzamide (Reference Example 31) in 60 ml of anhydrous N,N-dimethylformamide were added 1.51 g of (S)-(−)-3-(dimethylamino)pyrrolidine and 1.83 g of potassium carbonate, followed by stirring at room temperature for 14 hours. To the reaction solution were added water and an aqueous saturated sodium hydrogen carbonate solution, and the mixture was extracted with ethyl acetate and dried over anhydrous magnesium sulfate. The solvent was distilled off under reduced pressure and the residue was purified by silica gel column chromatography to obtain 4.57 g of pale yellow crystals.
Melting point: 179-183° C. (with decomposition)
A series of 3-substituted benzamide derivatives of STI-571 (imatinib mesylate) was prepared and evaluated for antiproliferative activity against the Bcr-Abl-positive leukemia cell line K562. Several 3-halogenated and 3-trifluoromethylated compounds, including NS-187, showed excellent potency.
Reagents and conditions: (a) NaBrO3, NaHSO3, EtOAc; (b) (COCl)2, cat. DMF, CH2Cl2, rt; (c) 7, K2CO3, dioxane, rt; (d) cyclic amines, K2CO3, DMF, rt.
………………………………
Bioorganic and Medicinal Chemistry Letters, 2007 , vol. 17, 10 pg. 2712 – 2717
|
References |
1: Peter B, Hadzijusufovic E, Blatt K, Gleixner KV, Pickl WF, Thaiwong T, Yuzbasiyan-Gurkan V, Willmann M, Valent P. KIT polymorphisms and mutations determine responses of neoplastic mast cells to bafetinib (INNO-406). Exp Hematol. 2010 Sep;38(9):782-91. doi: 10.1016/j.exphem.2010.05.004. Epub 2010 May 26. PubMed PMID: 20685234.
2: Kantarjian H, le Coutre P, Cortes J, Pinilla-Ibarz J, Nagler A, Hochhaus A, Kimura S, Ottmann O. Phase 1 study of INNO-406, a dual Abl/Lyn kinase inhibitor, in Philadelphia chromosome-positive leukemias after imatinib resistance or intolerance. Cancer. 2010 Jun 1;116(11):2665-72. doi: 10.1002/cncr.25079. PubMed PMID: 20310049; PubMed Central PMCID: PMC2876208.
3: Rix U, Remsing Rix LL, Terker AS, Fernbach NV, Hantschel O, Planyavsky M, Breitwieser FP, Herrmann H, Colinge J, Bennett KL, Augustin M, Till JH, Heinrich MC, Valent P, Superti-Furga G. A comprehensive target selectivity survey of the BCR-ABL kinase inhibitor INNO-406 by kinase profiling and chemical proteomics in chronic myeloid leukemia cells. Leukemia. 2010 Jan;24(1):44-50. doi: 10.1038/leu.2009.228. Epub 2009 Nov 5. PubMed PMID: 19890374.
4: Kamitsuji Y, Kuroda J, Kimura S, Toyokuni S, Watanabe K, Ashihara E, Tanaka H, Yui Y, Watanabe M, Matsubara H, Mizushima Y, Hiraumi Y, Kawata E, Yoshikawa T, Maekawa T, Nakahata T, Adachi S. The Bcr-Abl kinase inhibitor INNO-406 induces autophagy and different modes of cell death execution in Bcr-Abl-positive leukemias. Cell Death Differ. 2008 Nov;15(11):1712-22. doi: 10.1038/cdd.2008.107. Epub 2008 Jul 11. PubMed PMID: 18617896.
5: Morinaga K, Yamauchi T, Kimura S, Maekawa T, Ueda T. Overcoming imatinib resistance using Src inhibitor CGP76030, Abl inhibitor nilotinib and Abl/Lyn inhibitor INNO-406 in newly established K562 variants with BCR-ABL gene amplification. Int J Cancer. 2008 Jun 1;122(11):2621-7. doi: 10.1002/ijc.23435. PubMed PMID: 18338755.
6: Deguchi Y, Kimura S, Ashihara E, Niwa T, Hodohara K, Fujiyama Y, Maekawa T. Comparison of imatinib, dasatinib, nilotinib and INNO-406 in imatinib-resistant cell lines. Leuk Res. 2008 Jun;32(6):980-3. doi: 10.1016/j.leukres.2007.11.008. Epub 2008 Jan 8. PubMed PMID: 18191450.
7: Pan J, Quintás-Cardama A, Manshouri T, Cortes J, Kantarjian H, Verstovsek S. Sensitivity of human cells bearing oncogenic mutant kit isoforms to the novel tyrosine kinase inhibitor INNO-406. Cancer Sci. 2007 Aug;98(8):1223-5. Epub 2007 May 22. PubMed PMID: 17517053.
8: Kuroda J, Kimura S, Strasser A, Andreeff M, O’Reilly LA, Ashihara E, Kamitsuji Y, Yokota A, Kawata E, Takeuchi M, Tanaka R, Tabe Y, Taniwaki M, Maekawa T. Apoptosis-based dual molecular targeting by INNO-406, a second-generation Bcr-Abl inhibitor, and ABT-737, an inhibitor of antiapoptotic Bcl-2 proteins, against Bcr-Abl-positive leukemia. Cell Death Differ. 2007 Sep;14(9):1667-77. Epub 2007 May 18. PubMed PMID: 17510658.
9: Maekawa T. [Innovation of clinical trials for anti-cancer drugs in Japan–proposals from academia with special reference to the development of novel Bcr-Abl/Lyn tyrosine kinase inhibitor INNO-406 (NS-187) for imatinib-resistant chronic myelogenous leukemia]. Gan To Kagaku Ryoho. 2007 Feb;34(2):301-4. Japanese. PubMed PMID: 17301549.
10: Niwa T, Asaki T, Kimura S. NS-187 (INNO-406), a Bcr-Abl/Lyn dual tyrosine kinase inhibitor. Anal Chem Insights. 2007 Nov 14;2:93-106. PubMed PMID: 19662183; PubMed Central PMCID: PMC2716809.
11: Yokota A, Kimura S, Masuda S, Ashihara E, Kuroda J, Sato K, Kamitsuji Y, Kawata E, Deguchi Y, Urasaki Y, Terui Y, Ruthardt M, Ueda T, Hatake K, Inui K, Maekawa T. INNO-406, a novel BCR-ABL/Lyn dual tyrosine kinase inhibitor, suppresses the growth of Ph+ leukemia cells in the central nervous system, and cyclosporine A augments its in vivo activity. Blood. 2007 Jan 1;109(1):306-14. Epub 2006 Sep 5. PubMed PMID: 16954504.
Bafetinib in its binding site
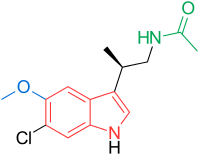
N-[(2R)-2-(6-chloro-5-methoxy-1H-indol-3-yl)propyl]acetamide
LY-156735 (TIK-301, PD-6735) is a melatonin MT1 and MT2 agonist which is under development for the treatment of insomnia and other sleep disorders.[1]
Beta-methyl-6-chloromelatonin (PD-6735) is a melatonin MT1 and MT2 agonist which had been in phase II trials at Phase 2 Discovery for the treatment of sleep latency in patients with primary insomnia, however, no recent development has been reported.
The melatonin agonist exhibits high selectivity and provides a novel mode of action different from that of benzodiazepine receptor ligands currently on the market.
Furthermore, the drug candidate is believed to be non-addicting, therefore, offering an advantage over marketed sleep medications. Originally discovered by Lilly, PD-6735 was licensed to Phase 2 Discovery in 2002 for further development.
Orphan drug designation has been assigned in the U.S. for the treatment of circadian rhythm sleep disorders in blind people with no light perception and for the treatment of neuroleptic-induced tardive dyskinesia in schizophrenia patients.
In 2007, the product candidate was licensed to Tikvah Therapeutics by Phase 2 Discovery for worldwide development and commercialization for the treatment of sleep disorder, depression and circadian rhythm disorder.
beta -alkylmelatonins as ovulation inhibitors [US4997845]1991-03-05
BETA-ALKYLMELATONINS [EP0281242]1988-09-07 GRANT1992-08-12

The condensation of 6-chloro-5-methoxy-1H-indole (I) with Meldrum’s acid (II) and acetaldehyde (III) catalyzed by L-proline in acetonitrile gives the adduct (IV), which is treated with Cu and ethanol in refluxing pyridine to yield 3-(6-chloro-5-methoxy-1H-indol-3-yl)butyric acid ethyl ester (V). The reaction of (V) with hydrazine at 140 C affords the hydrazide (VI), which is treated with NaNO2 and Ac-OH to provide the corresponding azide that, without isolation, is thermolyzed and rearranged in toluene at 80?C to give 7-chloro-6-methoxy-4-methyl-1,2,3,4-tetrahydro-9H-pyrido[3,4-b]indol-1-one (VII). The cleavage of the lactam ring of (VII) with KOH in refluxing ethanol/water yields 3-(2-amino-1-methylethyl)-6-chloro-5-methoxy-1H-indole-2-carboxylic acid (VIII). The decarboxylation of (VIII) by means of refluxing aq. 3M HCl affords 3-(2-amino-1-methylethyl)-6-chloro-5-methoxy-1H-indole (IX), which is finally acylated with acetic anhydride and pyridine in toluene to provide the target 6-chloromelatonin as a racemic compound.
EP 0281242;……….http://www.google.com/patents/EP0281242B1?cl=en
Example 3
…………………………………………….
PATENT
http://www.google.com/patents/EP0281242B1?cl=en

The intermediate diazonium salt (XIII) has been obtained as follows: the hydrogenation of 3-chloro-4-methoxynitrobenzene (XI) with H2 over Pt/Al2O3 in toluene gives the corresponding aniline (XII), which is diazotized with NaNO2/HCl and treated with sodium tetrafluoroborate to yield the target diazonium salt intermediate (XIII). The reduction of pulegone (I) with H2 over Pd/C gives the menthol (II), which is oxidized with CrO3/H2SO4 to yield 3(R),7-dimethyl-6-oxooctanoic acid (IV), which can also be obtained by direct oxidation of (l)-menthol (III) under the same conditions.
The oxidation of (IV) with trifluoroperacetic acid (trifluoroacetic anhydride/H2O2) in dichloromethane yields the 3(R)-methylhexanedioic acid isopropyl monoester (V), which is treated with NaOEt in ethanol to obtain the corresponding ethyl monoester (VI). The reaction of (VI) with diethyl carbonate, EtONa, and “Adogen 464″ (a phase transfer catalyst) in ethanol affords 5,5-bis(ethoxycarbonyl)-3(S)-methylpentanoic acid (VII), which is treated with oxalyl chloride to provide the expected acyl chloride (VIII). The reaction of (VIII) with sodium azide and benzyl alcohol gives the intermediate azide that rearranges to the benzyl carbamate (IX).
The reductive cyclization of (IX) with H2 over Pd/C in ethanol yields 5(R)-methyl-2-oxopiperidine-3-carboxylic acid ethyl ester (X), which is condensed with the intermediate diazonium salt (XIII) to afford the hydrazono derivative (XIV). The cyclization of (XIV) in hot formic acid provides 7-chloro-6-methoxy-4(R)-methyl-1,2,3,4-tetrahydro-9H-pyrido[3,4-b]indol-1-one (XV), which is treated with KOH In refluxing ethanol/water to cleave the lactam ring, yielding 3-(2-amino-1(R)-methylethyl)-6-chloro-5-methoxy-1H-indole-2-carboxylic acid (XVI). The decarboxylation of (XVI) by means of refluxing 3M HCl affords 3-(2-amino-1(R)-methylethyl)-6-chloro-5-methoxy-1H-indole (XVII), which is finally acylated with Ac2O and pyridine in toluene to provide the target 6-chloromelatonin as a pure enantiomer.
Example 7
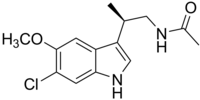 |
|
| Systematic (IUPAC) name | |
|---|---|
| N-[(2R)-(6-Chloro-5-methoxy-1H-indol-3-yl)propyl]acetamide | |
| Clinical data | |
| Legal status |
?
|
| Identifiers | |
| CAS number | 118702-11-7 |
| ATC code | ? |
| PubChem | CID 219018 |
| ChemSpider | 189853 |
| Chemical data | |
| Formula | C14H17ClN2O2 |
| Molecular mass | 280.757 |
SDVYCEVCEFLVKEVTKLIDNNKTEKEILDAFDKMCSKLPKSLSEECQEVVDTYGSSILSILLEEV SPELVCSMLHLCSG [SEQ ID NO: 2].
BXQ-350
Cincinnati Children’s Hospital ……..innovator
Bexion Pharmaceuticals……….under license
In February 2015, the US FDA granted saposin C Orphan designation for the treatment of glioblastoma multiforme
SAPOCIN C
Recombinant human Saposin C (SapC) bound to a liposomal formulation of the dioleoylphosphatidylserine
Bexion’s Saposin C – the active ingredient in the brain tumour therapy BXQ-350 – has been awarded Orphan Drug status by US regulators.
Read more at: http://www.pharmatimes.com/Article/15-02-17/US_Orphan_status_for_Bexion_s_brain_tumour_drug.aspx#ixzz3S3zXdHlO
Bexion Pharmaceuticals, under license from the Cincinnati Children’s Hospital, is investigating a human saposin C (SapC)/liposomal dioleoylphosphatidylserine (DOPS) conjugate, SapC-DOPS (BXQ-350), a nanovesicle-formulated pro-apoptotic sphingomyelinase activating molecular imaging agent and anticancer agent, for the potential diagnosis and treatment of cancer , . In October 2013, Bexion was planning a phase I first-in-human trial for the therapy of glioblastoma multiforme

Bexion Pharmaceuticals LLC announced today that the U.S. Food and Drug Administration (FDA) has granted the company Orphan Drug designation for Saposin C, active ingredient in its proprietary drug BXQ-350 for the potential treatment of glioblastoma multiforme.
The FDA’s Office of Orphan Drug Products Development reviews applications for Orphan Drug status to support development of medicines for underserved patient populations, or rare disorders that affect fewer than 200,000 people in the United States. The successful application submitted by Bexion and the FDA granting of Orphan Drug status entitles the company to a seven-year period of marketing exclusivity in the United States for BXQ-350, if it is approved by the FDA for the treatment of glioblastoma multiforme. Orphan Drug status also enables the company to apply for research grant funding for Phase I and II Clinical Trials, tax credits for certain research expenses, and a waiver from the FDA’s application user fee, as well as additional support from FDA and a potentially faster regulatory process.
Bexion was previously awarded a prestigious Phase II Bridge Award (Small Business Innovation Research Grant; SBIR) from the National Cancer Institute (NCI) to support the manufacture and clinical testing of BXQ-350.
“Orphan Drug status for BXQ-350 is an important milestone in the development of this new treatment modality,” stated Dr. Ray Takigiku, founder and CEO of Bexion. “Few treatment options are available for patients suffering from glioblastoma multiforme and this designation recognizes the unmet need that exists with this disease, as well as the unique attributes of BXQ-350. In addition, orphan designation allows Bexion to benefit from important financial, regulatory and commercial considerations and we have seen recently that products with orphan designation have become sought after assets.”

About Orphan Drug Designation
Orphan Drug designation is a status assigned to a medicine intended for use in rare diseases. In the U.S., the Orphan Drug Designation program confers Orphan Drug status to successful applicants for medicines intended for the safe and effective treatment, diagnosis or prevention of rare diseases or disorders that affect fewer than 200,000 people in the U.S. or that are not expected to recover the costs of developing and marketing a treatment.1
The approval of an orphan designation request does not alter the standard regulatory requirements and process for obtaining marketing approval for investigational use. Sponsors must establish safety and efficacy of a compound in the treatment of a disease through adequate and well-controlled studies. However, the FDA review process may be speedier for Orphan Drugs than those which do not receive Orphan Drug designation.
About BXQ-350
In pre-clinical studies, Bexion’s first-in-class biologic, BXQ-350 has shown promising results in selectively inducing cell death in the laboratory. BXQ-350 is a proprietary nanovesicle formulation of Saposin C (sphingolipid activator protein C, or SapC) and the phospholipid dioleoylphosphatidylserine (DOPS).
About Bexion Pharmaceuticals
Bexion Pharmaceuticals is a privately held biotech company focused on the development and commercialization of innovative cures for cancer. Initial products are based on a proprietary platform technology licensed from Cincinnati Children’s Hospital Medical Center. The technology has demonstrated potential for development as a therapeutic, diagnostic and surgical imaging reagent, and as a carrier for other pharmaceutical agents, such as oligonucleotides. For more information, visit www.bexionpharma.com or contact Margaret van Gilse atmvangilse@bexionpharma.com.
1 U.S. Food and Drug Administration web site. “Regulatory Information: Orphan Drug Act.”http://www.fda.gov/regulatoryinformation/legislation/federalfooddrugandcosmeticactfdcact/significantamendmentstothefdcact/orphandrugact/default.htm.
Margaret van Gilse859-757-1652mvangilse@bexionpharma.com
SOURCE Bexion Pharmaceuticals LLC
Glioblastoma is the most common primary CNS malignant neoplasm in adults, and accounts for nearly 75% of the cases. Although there has been steady progress in their treatment due to improvements in neuro-imaging, microsurgery, and radiation, glioblastomas remain incurable. The average life expectancy is less than one year from diagnosis, and the five-year survival rate following aggressive therapy, including gross tumor resection, is less than 10%. Glioblastomas cause death due to rapid, aggressive, and infiltrative growth in the brain. The infiltrative growth pattern is responsible for the un-resectable nature of these tumors. Glioblastomas are also relatively resistant to radiation and chemotherapy, and therefore post-treatment recurrence rates are high. In addition, the immune response to the neoplastic cells is mainly ineffective in completely eradicating residual neoplastic cells following resection and radiation therapy.
One problem in treating glioblastoma is the tumor’s protection behind the blood-brain tumor barrier (BBTB). A significant obstacle in the development of therapeutics for glioblastoma is the inability of systemic therapies to efficiently cross the BBTB. Saposin C (SapC) is a sphingolipid- activating protein that functions to catabolize glycosphingolipids. SapC-DOPS forms stable nanovesicles which can efficiently cross the blood-brain tumor barrier and fuse with GBM cells inducing cell death.
Rapamycin is a macrolide antibiotic produced by Streptomyces hygroscopicus, which was discovered first for its properties as an antifungal agent. Streptomyces hygroscopicus has also been implicated as a cancer agent.
There remains a need in the art for new therapeutics for the treatment of glioblastoma.
…………………………………………………………………..
https://www.google.com/patents/US20040229799?cl=en22
Example 1Purification of Recombinant Saposin C
[0106] Recombinant saposin C was overexpressed in E. coli cells by using the isopropyl-1-thio-β-D-galactopyranoside inducing pET system (Qi et al. (1994) J. Biol. Chem. 269:16746-16753, herein incorporated by reference in its entirety). Expressed polypeptides with a His-tag were eluted from nickel columns. After dialysis, the polypeptides were further purified by HPLC chromatography as follows. A C4 reverse phase column was equilibrated with 0.1% trifluoroacetic acid (TFA) for 10 minutes. The proteins were eluted in a linear (0-100%) gradient of 0.1% TFA in acetonitrile over 60 minutes. The major protein peak was collected and lyophilized. Protein concentration was determined as previously described (Qi et al. (1994) J. Biol. Chem. 269:16746-16753).
Example 2Bath Sonication of Sanosin C and Dioleoylphosphatidylserine
[0107] Dioleoylphosphatidylserine (DOPS) was obtained from Avanti Polar Lipids (Alabaster AL). Twenty to thirty imoles of DOPS in chloroform were dried under N2 and vacuum to lipid films. Five to ten μmoles saposin C polypeptide was added to the dried films and suspended in 50 μl McIlvanine buffer (pH 4.7). The suspension was then brought to a 1 ml volume with either cell culture medium or phosphate buffered saline (PBS) (Ausubel et al. (2002) Current Protocols in Molecular Biology. John Wiley & Sons, New York, New York, herein incorporated by reference). The mixture was sonicated in a bath sonicator for approximately 20 minutes. Ice was added as needed to prevent overheating the samples.

………………………………………………………………
http://www.google.com/patents/WO2014078522A1?cl=en
The SapC-DOPS composition comprises a phospholipid, an isolated saposin C-related polypeptide, wherein the polypeptide comprises an amino acid sequence at least 75% identical to the entire length of SEQ ID NO: 2, and a pharmaceutically acceptable carrier, wherein the phospholipid forms a nano vesicle incorporating the polypeptide. In certain embodiments, the polypeptide comprises an amino acid sequence at least 85% identical to the entire length of SEQ ID NO: 2. In certain embodiments, the polypeptide comprises an amino acid sequence at least 95% identical to the entire length of SEQ ID NO: 2. In certain embodiments, the polypeptide comprises an amino acid sequence at least 99% identical to the entire length of SEQ ID NO: 2.
The Sequence Listing, filed electronically and identified as SEQ_LIST_OSIF-2013- 102.txt, was created on November 12, 2013, is 5,548 in size, and is hereby incorporated by reference.
[0004] SEQ ID NO: 1
siy
J su c n 61y &n
*8 a 210 2iS
t n«
:?e
<H ¾■ yts ca« ¾»* **u v ΆΧ» s?s ass ¾«¾
:»o
L st S«x ri» r s
SEQ ID NO: 2

BEXION PHARMA



Givinostat (INN[1]) or gavinostat (originally ITF2357) is a histone deacetylase inhibitor with potential anti-inflammatory, anti-angiogenic, and antineoplastic activities.[2] It is a hydroxamate used in the form of its hydrochloride.
Givinostat is in numerous phase II clinical trials (including for relapsed leukemias and myelomas),[3] and has been granted orphan drug designation in the European Union for the treatment of systemic juvenile idiopathic arthritis[4] and polycythaemia vera.[5]
In 2010, orphan drug designation was assigned in the E.U. for the treatment of systemic-onset juvenile idiopathic arthritis and for the treatment of polycythemia vera. In 2013, this designation was assigned by the FDA for the treatment of Duchenne’s muscular dystrophy and for the treatment of Becker’s muscular dystrophy.
ITF2357 was discovered at Italfarmaco of Milan, Italy. It was patented in 1997 and first described in the scientific literature in 2005.[6][7]
Givinostat hydrochloride, an orally active, synthetic inhibitor of histone deacetylase, is being evaluated in several early clinical studies at Italfarmaco, including studies for the treatment of myeloproliferative diseases, polycythemia vera, Duchenne’s muscular dystrophy and periodic fever syndrome. The company was also conducting clinical trials for the treatment of Crohn’s disease and chronic lymphocytic leukemia; however, the trials were terminated.
No recent development has been reported for research into the treatment of juvenile rheumatoid arthritis, for the treatment of multiple myeloma and for the treatment of Hodgkin’s lymphoma.
Muscular dystrophies (MDs) include a heterogeneous group of genetic diseases invariably leading to muscle degeneration and impaired function. Mutation of nearly 30 genes gives rise to various forms of muscular dystrophy, which differ in age of onset, severity, and muscle groups affected (Dalkilic I, Kunkel LM. (2003) Muscular dystrophies: genes to pathogenesis. Curr. Opin. Genet. Dev. 13:231-238). The most common MD is the Duchenne muscular dystrophy (DMD), a severe recessive X-linked disease which affects one in 3,500 males, characterized by rapid progression of muscle degeneration, eventually leading to loss of ambulation and death within the second decade of life.
Attempts to replace or correct the mutated gene, by means of gene or cell therapy, might result in a definitive solution for muscular dystrophy, but this is not easy to achieve. Alternative strategies that prevent or delay muscle degeneration, reduce inflammation or promote muscle metabolism or regeneration might all benefit patients and, in the. future, synergize with gene or cell therapy. Steroids that reduce inflammation are currently the only therapeutic tool used in the majority of DMD patients (Cossu G, Sampaolesi M . (2007) New therapies for Duchenne muscular dystrophy: challenges, prospects and clinical trials. TRENDS Mol . Med. 13:520-526).
Diethyl- [ 6- ( 4-hydroxycarbamoyl-phenyl-carbamoyloxy- methyl ) -naphthalen-2-yl-methyl ] -ammonium chloride , which is described in WO 97/43251 (anhydrous form) and in WO 2004/065355 (monohydrate crystal form), herein both incorporated by reference, is an anti-inflammatory agent which is able to inhibit the synthesis of the majority of pro-inflammatory cytokines whilst sparing anti-inflammatory ones. Diethyl- [ 6- ( 4-hydroxycarbamoyl-phenyl-carbamoyloxy- methyl ) -naphthalen-2-yl-methyl ] -ammonium chloride is also known as ITF2357.
The monohydrate crystal form of diethyl- [ 6- ( 4- hydroxycarbamoyl-phenyl-carbamoyloxy-methy1 ) – naphthalen-2-yl-methyl ] -ammonium chloride is known as Givinostat .
Givinostat is being evaluated in several clinical studies, including studies for the treatment of myeloproliferative diseases, polycythemia vera, periodic fever syndrome, Crohn’s disease and systemic- onset juvenile idiopathic arthritis. Orphan drug designation was assigned in the E.U. for the treatment of systemic-onset juvenile idiopathic arthritis and for the treatment of polycythemia vera.
Givinostat has been recently found to act also as a Histone Deacetylase inhibitor (WO 2011/048514).
Histone deacetylases ( HDAC ) are a family of enzymes capable of removing the acetyl group bound to the lysine residues in the N-terminal portion of histones or in other proteins.
HDACs can be subdivided into four classes, on the basis of structural homologies. Class I HDACs (HDAC 1, 2, 3 and 8) are similar to the RPD3 yeast protein and are located in the cell nucleus. Class II HDACs (HDAC 4, 5, 6, 7, 9 and 10) are similar to the HDA1 yeast protein and are located both in the nucleus and in the cytoplasm. Class III HDACs are a structurally distinct form of NAD-dependent enzymes correlated with the SIR2 yeast protein. Class IV (HDAC 11) consists at the moment of a single enzyme having particular structural characteristics. The HDACs of classes I, II and IV are zinc enzymes and can be inhibited by various classes of molecule: hydroxamic acid derivatives, cyclic tetrapeptides , short-chain fatty acids, aminobenzamides , derivatives of electrophilic ketones, and the like. Class III HDACs are not inhibited by hydroxamic acids, and their inhibitors have structural characteristics different from those of the other classes .
The expression “histone deacetylase inhibitor” in relation to the present invention is to be understood as meaning any molecule of natural, recombinant or synthetic origin capable of inhibiting the activity of at least one of the enzymes classified as histone deacetylases of class I, class II or class IV.
Although HDAC inhibitors, as a class, are considered to be potentially useful as anti-tumor agents, it is worth to note that, till now, only two of them (Vorinostat and Romidepsin) have been approved as drugs for the cure of a single tumor form (Cutaneous T-cell lymphoma ) .
It is evident that the pharmaceutical properties of each HDAC inhibitor may be different and depend on the specific profile of inhibitory potency, relative to the diverse iso-enzymes as well as on the particular pharmacokinetic behaviour and tissue distribution.
Some HDAC inhibitors have been claimed to be potentially useful, in combination with other agents, for the treatment of DMD (WO 2003/033678, WO 2004/050076, Consalvi S. et al. Histone Deacetylase Inhibitors in the Treatment of Muscular Dystrophies: Epigenetic Drugs for Genetic Diseases. (2011) Mol. Med. 17 : 457-465 ) .
The potential therapeutic use of HDAC inhibitors in DMD may however be hampered by the possible harmful effects of these relatively toxic agents, especially when used for long-term therapies in paediatric patients .
Givinostat, as anti-inflammatory agent, has been already used in a phase II study in children with Systemic Onset Juvenile Idiopathic Arthritis; Givinostat administered at 1.5 mg/kg/day for twelve weeks achieved ACR Pedi 30, 50 and 70 improvement of approximately 70% (Vojinovic J, Nemanja D. (2011) HDAC Inhibition in Rheumatoid Arthritis and Juvenile Idiopathic Arthritis. Mol. Med 17:397-403) showing only a limited number of mild or moderate but short lasting, adverse effects.
To date more than 500 patients (including 29 children) have been treated with Givinostat. Repeated dose toxicity studies were carried out in dogs, rats and monkeys. Oral daily doses of the drug were administered up to nine consecutive months. The drug was well tolerated with no overt toxicity at high doses. The “no adverse effect levels” (NOAEL) ranged from 10 to 25 mg/kg/day depending on the animal species and the duration of treatment.
In juvenile animals Givinostat at 60 mg/kg/day did not affect the behavioural and physical development and reproductive performance of pups.
No genotoxic effect was detected for Givinostat in the mouse lymphoma assay and the chromosomal aberration assay in vitro and in the micronucleus test and UDS test in vivo.
| Patent | Submitted | Granted |
|---|---|---|
| Monohydrate hydrochloride of the 4-hydroxycarbamoyl-phenyl)-carbamic acid (6-diethylaminomethyl-naphtalen-2-yl) ester [US7329689] | 2005-11-03 | 2008-02-12 |
In clinical trials of givinostat as a salvage therapy for advanced Hodgkin’s lymphoma, the most common adverse reactions were fatigue (seen in 50% of participants), mild diarrhea or abdominal pain (40% of participants), moderate thrombocytopenia (decreased platelet counts, seen in one third of patients), and mild leukopenia (a decrease in white blood cell levels, seen in 30% of patients). One-fifth of patients experienced prolongation of the QT interval, a measure of electrical conduction in the heart, severe enough to warrant temporary suspension of treatment.[8]
Givinostat inhibits class I and class II histone deacetylases (HDACs) and several pro-inflammatory cytokines. This reduces expression of tumour necrosis factor (TNF), interleukin 1α and β, and interleukin 6.[7]
It also has activity against cells expressing JAK2(V617F), a mutated form of the janus kinase 2 (JAK2) enzyme that is implicated in the pathophysiology of many myeloproliferative diseases, including polycythaemia vera.[9][10] In patients with polycythaemia, the reduction of mutant JAK2 concentrations by givinostat is believed to slow down the abnormal growth of erythrocytes and ameliorate the symptoms of the disease.[5]
………………….

PATENT
https://www.google.com/patents/WO2004065355A1?cl=en
Hydrochloride of (6-diethylaminomethyl-naphthalen-2-yl)- methyl ester of (4-hydroxycarbamoylphenyl)-carbamic acid (II)
has been described in US patent 6,034,096 as a derivative of hydroxamic acid having anti-inflammatory and immunosuppressive activity, probably owing to the ability thereof to inhibit the production of pro-inflammatory cyto ines. This compound is obtained according to
Example 12 of the above-mentioned patent as an anhydrous, amorphous, hygroscopic, deliquescent solid which is difficult to handle.
crystalline form of monohydrous hydrochloride of
(6-diethylaminomethyl-naphthalen-2-yl)-methyl ester of
(4~hydroxycarbamoylphenyl)-carbamic acid (I).
This form is particularly advantageous from the industrial perspective because it is stable and simpler to handle than the anhydrous and amorphous form described above.
………………
PATENT
http://www.google.co.in/patents/US7329689
Hydrochloride of (6-diethylaminomethyl-naphthalen-2-yl)-methyl ester of (4-hydroxycarbamoylphenyl)-carbamic acid (II)
has been described in U.S. Pat. No. 6,034,096 as a derivative of hydroxamic acid having anti-inflammatory and immunosuppressive activity, probably owing to the ability thereof to inhibit the production of pro-inflammatory cytokines. This compound is obtained according to Example 12 of the above-mentioned patent as an anhydrous, amorphous, hygroscopic, deliquescent solid which is difficult to handle.
The 4-(6-diethylaminomethyl-naphthalen-2-ylmethoxycarbonylamino)-benzoic acid can be prepared as described in Example 12, point C, of U.S. Pat. No. 6,034,096.
The acid (1.22 kg, 3 moles) was suspended in THF (19 l) and the mixture was agitated under nitrogen over night at ambient temperature. The mixture was then cooled to 0° C. and thionyl chloride (0.657 l, 9 moles) was added slowly, still under nitrogen, with the temperature being maintained below 10° C. The reaction mixture was heated under reflux for 60 minutes, DMF (26 ml) was added and the mixture was further heated under reflux for 60 minutes.
The solvent was evaporated under vacuum, toluene was added to the residue and was then evaporated. This operation was repeated twice, then the residue was suspended in THF (11.5 l) and the mixture was cooled to 0° C.
The mixture was then poured into a cold solution of hydroxylamine (50% aq., 1.6 l, 264 moles) in 5.7 l of water. The mixture was then cooled to ambient temperature and agitated for 30 minutes. 6M HCl was added until pH 2 was reached and the mixture was partially evaporated under vacuum in order to eliminate most of the THF. The solid was filtered, washed repeatedly with water and dissolved in a solution of sodium bicarbonate (2.5%, 12.2 l). The solution was extracted with 18.6 l of a mixture of THF and ethyl acetate (2:1 v/v). 37% HCl (130 ml) were added to the organic layer in order to precipitate the monohydrate of the (6-diethylaminomethyl-naphthalen-2-yl)-methyl ester hydrochloride of the (4-hydroxycarbamoyl-phenyl)-carbamic acid. If necessary, this operation can be repeated several times to remove any residues of the original acid.
Finally, the solid was dried under vacuum (approximately 30 mbar, 50° C.), producing 0.85 kg (60%) of compound (I).
HPLC purity: 99.5%; water content (Karl Fischer method): 3.8%; (argentometric) assay: 99.8%.
| Elemental analysis | |||||
| C % | H % | Cl % | N % | ||
| Calculated for | 60.56 | 6.35 | 7.45 | 8.83 | |
| C24H30ClN3O5 | |||||
| Found | 61.06 | 6.48 | 7.48 | 8.90 | |
PATENT
http://www.google.co.in/patents/US20120302633
…………………..
http://www.google.com/patents/US6034096
A. 1-(3-Dimethylaminopropyl)-3-ethylcarbodiimide hydrochloride (EDCI) (22.2 g, 115 mmol) was added to a solution of 2,6-naphthalenedicarboxylic acid (25 g, 115 mmol) and hydroxybenzotriazole (15.6 g, 115 mmol) in dimethylformamide (1800 ml) and the mixture was stirred at room temperature for 2 hours. Diethyl amine (34.3 ml, 345 mmol) was added and the solution was stirred overnight at room temperature. The solvent was then evaporated under reduced pressure and the crude was treated with 1N HCl (500 ml) and ethyl acetate (500 ml), insoluble compounds were filtered off and the phases were separated. The organic phase was extracted with 5% sodium carbonate (3×200 ml) and the combined aqueous solutions were acidified with concentrated HCl and extracted with ethyl acetate (3×200 ml). The organic solution was then washed with 1N HCl (6×100 ml), dried over anhydrous sodium sulphate and the solvent was removed under reduced pressure yielding 18.5 g (Yield 60%) of pure 6-(diethylaminocarbonyl)-2-naphthalenecarboxylic acid; m.p.=122-124° C.
1 H-NMR d 8.67 (s, 1H), 8.25-8.00 (m, 4H), 7.56 (d, 1H), 3.60-3.20 (m, 4H), 1.30-1.00 (m, 6H).
B. A solution of 6-(diethylaminocarbonyl)-2- naphthalenecarboxylic acid (18 g, 66 mmol) in THF (200 ml) was slowly added to a refluxing suspension of lithium aluminium hydride (7.5 g, 199 mmol) in THF (500 ml). The mixture was refluxed for an hour, then cooled at room temperature and treated with a mixture of THF (25 ml) and water (3.5 ml), with 20% sodium hydroxide (8.5 ml) and finally with water (33 ml). The white solid was filtered off and the solvent was removed under reduced pressure. Crude was dissolved in diethyl ether (200 ml) and extracted with 1N HCl (3×100 ml). The aqueous solution was treated with 32% sodium hydroxide and extracted with diethyl ether (3×100 ml). The organic solution was dried over anhydrous sodium sulphate and the solvent was removed under reduced pressure yielding 12.7 g (79% yield) of pure 6-(diethylaminomethyl)-2-naphthalenemethanol as thick oil.
1 H-NMR d 7.90-7.74 (m, 4H), 7.49 (m, 2H), 5.32 (t, 1H, exchange with D2 O), 4.68 (d, 2H), 3.69 (s, 2H), 2.52 (q, 4H), 1.01 (t, 6H).
C. A solution of 6-(diethylaminomethyl)-2-naphthalene-methanol (12.5 g, 51 mmol) and N,N’-disuccinimidyl carbonate (13.2 g, 51 mmol) in acetonitrile (250 ml) was stirred at room temperature for 3 hours, then the solvent was removed and the crude was dissolved in THF (110 ml). This solution was added to a solution of 4-amino benzoic acid (7.1 g, 51 mmol) and sodium carbonate (5.5 g, 51 mmol) in water (200 ml) and THF (100 ml). The mixture was stirred overnight at room temperature, then THF was removed under reduced pressure and the solution was treated with 1N HCl (102 ml, 102 mmol). The precipitate was filtered, dried under reduced pressure, tritured in diethyl ether and filtered yielding 13.2 g (yield 64%) of pure 4-[6-(diethylaminomethyl)naphth-2-ylmethyloxycarbamoyl]-benzoic acid; m.p.=201-205° C. (dec.)
1 H-NMR d 10.26 (s, 1H), 8.13 (s, 1H), 8.05-7.75 (m, 6H), 7.63 (m, 3H), 5.40 (s, 2H), 4.32 (s, 2H), 2.98 (q, 4H), 1.24 (t, 6H).
D. A solution of 4-[6-(diethylaminomethyl)naphth-2-ylmethyloxycarbamoyl]benzoic acid (13.1 g, 32 mmol) and thionyl chloride (7 ml, 96 mmol) in chloroform (300 ml) was refluxed for 4 hours, then the solvent and thionyl chloride were evaporated. Crude was dissolved in chloroform (100 ml) and evaporated to dryness three times. Crude was added as solid to a solution of hydroxylamine hydrochloride (2.7 g, 39 mmol) and sodium bicarbonate (5.4 g, 64 mmol) and 1N sodium hydroxide (39 ml, 39 mmol) in water (150 ml) and THF (50 ml). The mixture was stirred overnight at room temperature, then THF was removed under reduced pressure and the aqueous phase was extracted with ethyl acetate (3×100 ml). The combined organic phases were dried over anhydrous sodium sulphate and the solvent was removed under reduced pressure. Crude was dissolved in THF and treated with a 1.5 N etheric solution of HCl. The solid product was filtered and dried yielding 6 g (yield 41%) of pure 4-[6-(diethylaminomethyl)naphth-2-ylmethyloxycarbamoyl]benzohydroxamic acid hydrochloride as white solid; m.p.=162-165° C., (dec.)
1 H-NMR d 11.24 (s, 1H, exchange with D2 O), 10.88 (s, 1H, exchange with D2 O), 10.16 (s, 1H), 8.98 (bs, 1H, exchange with D2 O), 8.21 (s, 1H), 8.10-7.97 (m, 3H), 7.89 (d, 1H), 7.80-7.55 (m, 5H), 5.39 (s, 2H), 4.48 (d, 2H), 3.09 (m, 4H), 1.30 (t, 6H).

Some nmr predictions
CAS NO. 497833-27-9, [6-(diethylaminomethyl)naphthalen-2-yl]methyl N-[4-(hydroxycarbamoyl)phenyl]carbamate H-NMR spectral analysis
![[6-(diethylaminomethyl)naphthalen-2-yl]methyl N-[4-(hydroxycarbamoyl)phenyl]carbamate NMR spectra analysis, Chemical CAS NO. 497833-27-9 NMR spectral analysis, [6-(diethylaminomethyl)naphthalen-2-yl]methyl N-[4-(hydroxycarbamoyl)phenyl]carbamate H-NMR spectrum](http://pic11.molbase.net/nmr/nmr_image/2015-01-20/001/566/1566912_1h.png)
13 C NMR PREDICTIONS
![[6-(diethylaminomethyl)naphthalen-2-yl]methyl N-[4-(hydroxycarbamoyl)phenyl]carbamate NMR spectra analysis, Chemical CAS NO. 497833-27-9 NMR spectral analysis, [6-(diethylaminomethyl)naphthalen-2-yl]methyl N-[4-(hydroxycarbamoyl)phenyl]carbamate C-NMR spectrum](http://pic11.molbase.net/nmr/nmr_image/2015-01-20/001/566/1566912_13c.png)

COSY NMR…..http://www.nmrdb.org/

HMBC /HSQC

| US6034096 | 12 May 1997 | 7 Mar 2000 | Italfarmaco S.P.A. | Compounds with anti-inflammatory and immunosuppressive activities |
| WO1997043251A1 | May 12, 1997 | Nov 20, 1997 | Italfarmaco Spa | Compounds with anti-inflammatory and immunosuppressive activities |
| WO2004063146A1 | Jan 7, 2004 | Jul 29, 2004 | Italfarmaco Spa | Hydroxamic acid derivatives having anti-inflammatory action |
| WO2004065355A1 | Jan 8, 2004 | Aug 5, 2004 | Italfarmaco Spa | Monohydrate hydrochloride of the 4-hydroxycarbamoyl-phenyl)-carbamic acid (6-diethylaminomethyl-naphtalen-2-yl) ester |
| WO2006003068A2 | Jun 7, 2005 | Jan 12, 2006 | Italfarmaco Spa | Alpha-amino acid derivatives with antiinflammatory activity |
| WO2008097654A1 | Feb 8, 2008 | Aug 14, 2008 | Nancie M Archin | Methods of using saha for treating hiv infection |
| Citing Patent | Filing date | Publication date | Applicant | Title |
|---|---|---|---|---|
| US8518988 * | 3 Dec 2010 | 27 Aug 2013 | Chemi Spa | Polymorph of the hydrochloride of the (4-hydroxycarbamoyl-phenyl)-carbamic acid (6-dimethylamino methyl-2-naphthalenyl) ester |
| US20120302633 * | 3 Dec 2010 | 29 Nov 2012 | Chemi Spa | Novel polymorph of the hydrochloride of the (4-hydroxycarbamoyl-phenyl)-carbamic acid (6-dimethylamino methyl-2-naphthalenyl) ester |
| WO2011092556A1 | 3 Dec 2010 | 4 Aug 2011 | Chemi Spa | Novel polymorph of the hydrochloride of the (4-hydroxycarbamoyl-phenyl)-carbamic acid (6-dimethylamino methyl-2-naphtalenyl) ester |
 |
|
| Systematic (IUPAC) name | |
|---|---|
| {6-[(diethylamino)methyl]naphthalen-2-yl}methyl [4-(hydroxycarbamoyl)phenyl]carbamate | |
| Clinical data | |
|
|
| Legal status |
|
| Routes | Oral |
| Identifiers | |
| CAS number | 497833-27-9 |
| ATC code | None |
| PubChem | CID 9804992 |
| ChemSpider | 7980752 |
| UNII | 5P60F84FBH |
| Chemical data | |
| Formula | C24H27N3O4 |
| Molecular mass | 421.489 g/mol |
| Italfarmaco S.p.A. | |
|---|---|
 |
|
| Stato |  Italia Italia |
| Tipo | Società per azioni |
| Fondazione | 1938 a Milano |
| Fondata da | Gastone De Santis |
| Sede principale | Milano |
| Filiali |  Spagna – Spagna –  Portogallo Portogallo Grecia – Grecia –  Russia Russia Cile – Cile –  Brasile Brasile Turchia Turchia |
| Persone chiave | Francesco De Santis, [Presidente Holding] |
| Settore | sanità |
| Prodotti | Farmaci |
| Fatturato | >500 milioni di Euro (gruppo) (2011) |
| Dipendenti | >1900 (gruppo) (2011) |
| Sito web | www.italfarmaco.com |
MILAN ITALY


ELIGLUSTAT TARTRATE
THERAPEUTIC CLAIM Treatment of lysosomal storage disorders
CHEMICAL NAMES
1. Octanamide, N-[(1R,2R)-2-(2,3-dihydro-1,4-benzodioxin-6-yl)-2-hydroxy-1-(1-
pyrrolidinylmethyl)ethyl]-, (2R,3R)-2,3-dihydroxybutanedioate (2:1)
2. bis{N-[(1R,2R)-2-(2,3-dihydro-1,4-benzodioxin-6-yl)-2-hydroxy-1-(pyrrolidin-1-
ylmethyl)ethyl]octanamide} (2R,3R)-2,3-dihydroxybutanedioate
MOLECULAR FORMULA C23H36N2O4 . ½ C4H6O6
MOLECULAR WEIGHT 479.6
MANUFACTURER Genzyme Corp.
CODE DESIGNATION Genz-112638
CAS REGISTRY NUMBER 928659-70-5
Eliglustat (INN, USAN;[1] trade name Cerdelga) is a treatment for Gaucher’s disease developed by Genzyme Corp that was approved by the FDA August 2014.[2] Commonly used as the tartrate salt, the compound is believed to work by inhibition ofglucosylceramide synthase.[3][4]
In March 2015, eliglustat tartrate was approved in Japan for the treatment of Gaucher disease. Eliglustat tartrate was described specifically within the US FDA’s Orange Booked listed US6916802, which is set to expire in April 2022.
In May 2015, the Orange Book also listed that eliglustat tartrate had Orphan Drug Exclusivity and New Chemical Entity exclusivity until 2019 and 2021, respectively.
it having been developed and launched as eliglustat tartrate by Genzyme (a wholly owned subsidiary of Sanofi), under license from the University of Michigan.
Eliglustat tartrate is known to act as inhibitors of glucosylceramide synthase and glycolipid, useful for the treatment of Gaucher’s disease type I and lysosome storage disease.
What is Eliglustat?

Eliglustat tartrate (Genz-1 12638) is a glucocerebroside (glucosylceramide) synthase inhibitor for the treatment of gaucher disease and other lysosomal storage disorders, which is currently under development.
Eliglustat is chemically known as 1 R, 2R-Octanoic acid [2-(2′, 3′-dihydro-benzo [1 , 4] dioxin-6′-yl)-2-hydroxy-1 -pyrrolidin-1 -ylmethyl]-ethyl]-amide, having a structural formula I depicted here under.
Formula I
Eliglustat hemitartrate (Genz-1 12638) development by Genzyme, is a glucocerebroside (glucosylceramide) synthase inhibitor for the treatment of Gaucher disease and other lysosomal storage disorders. Eliglustat hemitartrate is orally active with potent effects on the primary identified molecular target for type 1 Gaucher disease and other glycosphingolipidoses, appears likely to fulfill high expectations for clinical efficacy.
Gaucher disease belongs to the class of lysosomal diseases known as glycosphingolipidoses, which result directly or indirectly from the accumulation of glycosphingolipids, many hundreds of which are derived from glucocerebroside. The first step in glycosphingolipid biosynthesis is the formation of glucocerebroside, the primary storage molecule in Gaucher disease, via glucocerebroside synthase (uridine diphosphate [UDP] – glucosylceramide glucosyl transferase). Eliglustat hemitartrate is based on improved inhibitors of glucocerebroside synthase.
U.S. patent No. 7,196,205 (herein described as US’205) discloses a process for the preparation of eliglustat or a pharmaceutically acceptable salt thereof. In this patent, eliglustat was synthesized via a seven-step process involving steps in that sequence:
(i) coupling S-(+)-2-phenyl glycinol with phenyl bromoacetate followed by column chromatography for purification of the resulting intermediate,
(ii) reacting the resulting (5S)-5-phenylmorpholin-2-one with 1 , 4-benzodioxan-6-carboxaldehyde to obtain a lactone,
(iii) opening the lactone of the oxazolo-oxazinone cyclo adduct via reaction with pyrrolidine,
(iv) hydrolyzing the oxazolidine ring, (v) reducing the amide to amine to obtain sphingosine like compound, (vi) reacting the resulting amine with octanoic acid and N-hydroxysuccinimide to obtain crude eliglustat, (vii) purifying the crude eliglustat by repeated isolation for four times from a mixture of ethyl acetate and n-heptane.
U.S. patent No. 6855830, 7265228, 7615573, 7763738, 8138353, U.S. patent application publication No. 2012/296088 disclose processes for preparation of eliglustat and intermediates thereof.
U.S. patent application publication No. 2013/137743 discloses (i) a hemitartrate salt of eliglustat, (ii) a hemitartrate salt of eliglustat, wherein at least 70% by weight of the salt is crystalline, (iii) a hemitartrate salt of Eliglustat, wherein at least 99% by weight of the salt is in a single crystalline form.
WO 2015059679
| Process for the preparation of eliglustat free base – comprising the reaction of S-(+)-phenyl glycinol with phenyl-alpha-bromoacetate to obtain 5-phenylmorpholin-2-one, which is further converted to eliglustat. | |
| Dr Reddy’s Laboratories Ltd | |
| New crystalline eliglustat free base Form R1 and a process for its preparation are claimed. Also claimed is a process for the preparation of eliglustat free base which comprises the reaction of S-(+)-phenyl glycinol with phenyl-alpha-bromoacetate to obtain 5-phenylmorpholin-2-one, which is further converted to eliglustat.Further eliglustat oxalate, its crystalline form, and a process for the preparation of crystalline eliglustat oxalate, are claimed. | |
Eliglustat tartrate (Genz-1 12638) is a glucocerebroside (glucosylceramide) synthase inhibitor for the treatment of gaucher disease and other lysosomal storage disorders, which is currently under development.
Eliglustat is chemically known as 1 R, 2R-Octanoic acid [2-(2′, 3′-dihydro-benzo [1 , 4] dioxin-6′-yl)-2-hydroxy-1 -pyrrolidin-1 -ylmethyl]-ethyl]-amide, having a structural formula I depicted here under.

Formula I
Eliglustat hemitartrate (Genz-1 12638) development by Genzyme, is a glucocerebroside (glucosylceramide) synthase inhibitor for the treatment of Gaucher disease and other lysosomal storage disorders. Eliglustat hemitartrate is orally active with potent effects on the primary identified molecular target for type 1 Gaucher disease and other glycosphingolipidoses, appears likely to fulfill high expectations for clinical efficacy.
Gaucher disease belongs to the class of lysosomal diseases known as glycosphingolipidoses, which result directly or indirectly from the accumulation of glycosphingolipids, many hundreds of which are derived from glucocerebroside. The first step in glycosphingolipid biosynthesis is the formation of glucocerebroside, the primary storage molecule in Gaucher disease, via glucocerebroside synthase (uridine diphosphate [UDP] – glucosylceramide glucosyl transferase). Eliglustat hemitartrate is based on improved inhibitors of glucocerebroside synthase.
U.S. patent No. 7,196,205 (herein described as US’205) discloses a process for the preparation of eliglustat or a pharmaceutically acceptable salt thereof. In this patent, eliglustat was synthesized via a seven-step process involving steps in that sequence:
(i) coupling S-(+)-2-phenyl glycinol with phenyl bromoacetate followed by column chromatography for purification of the resulting intermediate,
(ii) reacting the resulting (5S)-5-phenylmorpholin-2-one with 1 , 4-benzodioxan-6-carboxaldehyde to obtain a lactone,
(iii) opening the lactone of the oxazolo-oxazinone cyclo adduct via reaction with pyrrolidine,
(iv) hydrolyzing the oxazolidine ring, (v) reducing the amide to amine to obtain sphingosine like compound, (vi) reacting the resulting amine with octanoic acid and N-hydroxysuccinimide to obtain crude eliglustat, (vii) purifying the crude eliglustat by repeated isolation for four times from a mixture of ethyl acetate and n-heptane.
U.S. patent No. 6855830, 7265228, 7615573, 7763738, 8138353, U.S. patent application publication No. 2012/296088 disclose processes for preparation of eliglustat and intermediates thereof.
U.S. patent application publication No. 2013/137743 discloses (i) a hemitartrate salt of eliglustat, (ii) a hemitartrate salt of eliglustat, wherein at least 70% by weight of the salt is crystalline, (iii) a hemitartrate salt of Eliglustat, wherein at least 99% by weight of the salt is in a single crystalline form.
Example 1 : Preparation of 5-phenyl morpholine-2-one hydrochloride
To a (S) + phenyl glycinol (100g) add N, N-diisopropylethylamine (314ml) and acetonitrile (2000ml) under nitrogen atmosphere at room temperature. It was cooled to 10- 15° C. Phenyl bromoacetate (172.4g) dissolved in acetonitrile (500ml) was added to the above solution at 15° C over a period of 30 min. The reaction mixture is allowed to room temperature and stirred for 16-20h. Progress of the reaction was monitored by thin layer chromatography. After completion of the reaction, the reaction mixture was concentrated under reduced pressure at a water bath
temperature less than 25° C to get a residue. The residue was dissolved in ethyl acetate (1000ml) and stirred for 1 h at 15-20°C to obtain a white solid. The solid material obtained was filtered and washed with ethyl acetate (200ml). The filtrate was dried over anhydrous sodium sulphate (20g) and concentrated under reduced pressure at a water bath temperature less than 25° C to give crude compound (1000g) as brown syrup. The Crude brown syrup is converted to HCI salt by using HCI in ethyl acetate to afford 5-phenyl morpholine-2-one hydrochloride (44g) as a white solid. Yield: 50%, Mass: m/z = 177.6; HPLC (% Area Method): 90.5%
Example 2: Preparation of (1 R,3S,5S,8aS)-1 ,3-Bis-(2′,3′-dihydro-benzo[1 ,4] dioxin-6′-yl)-5-phenyl-tetrahydro-oxazolo[4,3-c][1 ,4]oxazin-8-one.
5-phenyl morpholine-2-one hydrochloride (100g) obtained from above stage 1 is dissolved in toluene (2500ml) under nitrogen atmosphere at 25-30°C. 1 ,4-benzodioxane-6-carboxaldehyde (185.3g) and sodium sulphate (400g) was added to the above solution and the reaction mixture was heated at 100-105°C for 72h. Progress of the reaction was monitored by thin layer chromatography. After completion of reaction, the reaction mixture was concentrated under reduced pressure at a water bath temperature less than 25° C to get a residue. The residue was cooled to 10°C, ethyl acetate (2700ml) and 50% sodium bisulphate solution (1351 ml) was added to the residue and stirred for 1 h at 10°C to obtain a white solid. The obtained white solid was filtered and washed with ethyl acetate. The separated ethyl acetate layer was washed with water (1000ml), brine (1000ml) and dried over anhydrous sodium sulphate. The organic layer was concentrated under reduced pressure at a water bath temperature of 45-50°C to get a crude material. The obtained crude material is triturated with diethyl ether (1500ml) to get a solid material which is filtered and dried under vacuum at room temperature for 2-3h to afford (1 R,3S,5S,8aS)-1 ,3-Bis-(2′,3′-dihydro-benzo[1 ,4]dioxin-6′-yl)-5-phenyl-tetrahydro-oxazolo[4,3-c][1 ,4]oxazin-8-one (148g) as a yellow solid. Yield: 54%, Mass: m/z = 487.7; HPLC (% Area Method): 95.4 %
Example 3: Preparation of (2S,3R,1 “S)-3-(2′,3′-(Dihydro-benzo[1 ,4]dioxin-6′-yl)-3-hydroxy-2-(2″-hydroxy-1 ”^henyl-ethy^
(1 R,3S,5S,8aS)-1 !3-Bis-(2′!3′-dihydro-benzo[1 ,4]dioxin-6′-yl)-5-phenyl-tetrahydro-oxazolo[4,3-c][1 ,4]oxazin-8-one (70g) obtained from above stage 2 was dissolved in chloroform (1400ml) at room temperature. It was cooled to 0-5°C and pyrrolidone (59.5ml) was added at 0-5°C over a period of 30 minutes. The reaction mixture was allowed to room temperature and stirred for 16-18h. Progress of the reaction was monitored by thin layer chromatography. After completion of reaction, the reaction mixture was concentrated under reduced pressure at a water bath temperature of 40-45°C to obtain a crude. The obtained crude was dissolved in methanol (1190ml) and 1 N HCI (1 190ml) at 10-15° C, stirred for 10 minutes and heated at 80-85°C for 7h. Progress of the reaction was monitored by thin layer chromatography. After completion of reaction, methanol was concentrated under reduced pressure at a water bath temperature of 50-55°C.The aqueous layer was extracted with ethyl acetate and the organic layer was washed with 1 N HCI (50ml). The aqueous layer was basified with saturated sodium bicarbonate solution up to pH 8-9 and extracted with ethyl acetate (3x70ml). The combined organic layers was washed with brine (100ml), dried over anhydrous sodium sulphate and concentrated under reduced pressure at a water bath temperature of 50-55°C to afford (2S,3R,1″S)-3-(2′,3′-(Dihydro-benzo[1 ,4]dioxin-6′-yl)-3-hydroxy-2-(2″-hydroxy-1 “-phenyl-ethylamino)-1 -pyrrolidin-1 -yl-propan-1 -one (53g) as a yellow foamy solid. Yield: 90%, Mass: m/z = 412.7, HPLC (% Area Method): 85.1 %
Example 4: Preparation of (1 R,2R,1 “S)-1-(2′,3′-(Dihydro-benzo[1 ,4]dioxin-6′-yl)2-hydroxy-2-(2″-hydroxy-1 ‘-phenyl-ethylamino)-3-pyrrolidin-1-yl-propan-1-ol.
(2S,3R,1 “S)-3-(2′,3′-(Dihydro-benzo[1 ,4]dioxin-6′-yl)-3-hydroxy-2-(2″-hydroxy-1 “-phenyl-ethylamino)-1 -pyrrolidin-1 -yl-propan-1 -one (2.5g) obtained from above stage 3 dissolved in Tetrahydrofuran (106ml) was added to a solution of Lithium aluminium hydride (12.2g) in tetrahydrofuran (795ml) at 0°C and the reaction mixture was heated at 60-65°C for 10h. Progress of the reaction was monitored by thin layer chromatography. After completion of reaction, the reaction mixture was cooled to 5- 10°C and quenched in saturated sodium sulphate solution (100ml) at 5-10°C. Ethyl acetate was added to the reaction mass and stirred for 30-45 min. The obtained solid is filtered through celite bed and washed with ethyl acetate. Filtrate was dried over anhydrous sodium sulphate and concentrated under reduced pressure at a water bath temperature of 50°C to afford (1 R,2R, 1″S)-1 -(2′,3′-(Dihydro-benzo[1 ,4]dioxin-6′-yl)2-hydroxy-2-(2″-hydroxy-1 ‘-phenyl-ethylamino)-3-pyrrolidin-1 -yl-propan-1 -ol (43.51 g) as a yellow gummy liquid. The crude is used for the next step without further purification. Yield: 85%, Mass: m/z = 398.7, HPLC (% Area Method): 77 %
Example 5: Preparation of (1 R, 2R)-2-Amino-1-(2′, 3′-dihydro-benzo [1 , 4] dioxin-6′-yl)-3-pyrrolidin-1 -yl-propan-1 -ol.
(1 R,2R,1 “S)-1 -(2′,3′-(Dihydro-benzo[1 ,4]dioxin-6′-yl)2-hydroxy-2-(2″-hydroxy-1 ‘-phenyl-ethylamino)-3-pyrrolidin-1 -yl-propan-1 -ol (40g) obtained from above stage 4 was dissolved in methanol (400ml) at room temperature in a 2L hydrogenation flask. Trifluoroacetic acid (15.5ml) and 20% Pd (OH) 2 (40g) was added to the above solution under nitrogen atmosphere. The reaction mixture was hydrogenated under H2, 10Opsi for 16-18h at room temperature. Progress of the reaction was monitored by thin layer chromatography. After completion of reaction, the reaction mixture was filtered through celite bed and washed with methanol (44ml) and water (44ml). Methanol was concentrated under reduced pressure at a water bath temperature of 50-55°C and the aqueous layer was washed with ethyl acetate. The aqueous layer was basified with 10M NaOH till the PH reaches 12-14 and then extracted with dichloromethane (2x125ml). The organic layer was dried over anhydrous sodium sulphate (3gm) and concentrated under reduced pressure at a water bath temperature of 45°C to obtain a gummy liquid. The gummy liquid was triturated with methyl tertiary butyl ether for 1 h to get a white solid, which is filtered and dried under vacuum at room temperature to afford (1 R, 2R)-2-Amino-1 -(2′, 3′-dihydro-benzo [1 , 4] dioxin-6′-yl)-3-pyrrolidin-1 -yl-propan-1 -ol (23g) as a white solid. Yield: 82.3%, Mass (m/zj: 278.8, HPLC (% Area Method): 99.5%, Chiral HPLC (% Area Method): 97.9%
Example 6: Preparation of Eliglustat {(1 R, 2R)-Octanoic acid[2-(2′,3′-dihydro-benzo [1 , 4] dioxin-6′-yl)-2-hydroxy-1 -pyrrolidin-1-ylmethyl-ethyl]-amide}.
(1 R, 2R)-2-Amino-1 -(2′, 3′-dihydro-benzo [1 , 4] dioxin-6′-yl)-3-pyrrolidin-1 -yl-propan-1 -ol (15g) obtained from above stage 5 was dissolved in dry dichloromethane (150ml) at room temperature under nitrogen atmosphere and cooled to 10-15° C. Octanoic acid N-hydroxy succinimide ester (13.0 g)was added to the above reaction mass at 10-15° C and stirred for 15 min. The reaction mixture was stirred at room temperature for 16h-18h. Progress of the reaction was monitored by thin layer chromatography. After completion of reaction, the reaction mixture was cooled to 15°C and diluted with 2M NaOH solution (100 ml_) and stirred for 20 min at 20 °C. The organic layer was separated and washed with 2M sodium hydroxide (3x90ml).The organic layer was dried over anhydrous sodium sulphate (30g) and concentrated under reduced pressure at a water bath temperature of 45°C to give the crude compound (20g).The crude is again dissolved in methyl tertiary butyl ether (25 ml_) and precipitated with Hexane (60ml). It is stirred for 10 min, filtered and dried under vacuum to afford Eliglustat as a white solid (16g). Yield: 74%, Mass (m/zj: 404.7 HPLC (% Area Method): 97.5 %, ELSD (% Area Method): 99.78%, Chiral HPLC (% Area Method): 99.78 %.
Example 7: Preparation of Eliglustat oxalate.
Eliglustat (5g) obtained from above stage 6 is dissolved in Ethyl acetate (5ml) at room temperature under nitrogen atmosphere. Oxalic acid (2.22g) dissolved in ethyl acetate (5ml) was added to the above solution at room temperature and stirred for 14h. White solid observed in the reaction mixture was filtered and dried under vacuum at room temperature for 1 h to afford Eliglustat oxalate as a white solid (4g). Yield: 65.46%, Mass (m/zj: 404.8 [M+H] +> HPLC (% Area Method): 95.52 %, Chiral HPLC (% Area Method): 99.86 %
……………………………..
Nmr predict
![N-[(1R,2R)-1-(2,3-dihydro-1,4-benzodioxin-6-yl)-1-hydroxy-3-pyrrolidin-1-ylpropan-2-yl]octanamide NMR spectra analysis, Chemical CAS NO. 491833-29-5 NMR spectral analysis, N-[(1R,2R)-1-(2,3-dihydro-1,4-benzodioxin-6-yl)-1-hydroxy-3-pyrrolidin-1-ylpropan-2-yl]octanamide H-NMR spectrum](http://pic11.molbase.net/nmr/nmr_image/2014-09-06/001/571/702/491833-29-5-1h.png)
13 C NMR
![N-[(1R,2R)-1-(2,3-dihydro-1,4-benzodioxin-6-yl)-1-hydroxy-3-pyrrolidin-1-ylpropan-2-yl]octanamide NMR spectra analysis, Chemical CAS NO. 491833-29-5 NMR spectral analysis, N-[(1R,2R)-1-(2,3-dihydro-1,4-benzodioxin-6-yl)-1-hydroxy-3-pyrrolidin-1-ylpropan-2-yl]octanamide C-NMR spectrum](http://pic11.molbase.net/nmr/nmr_image/2014-09-06/001/571/702/491833-29-5-13c.png)
CAS NO. 491833-29-5, N-[(1R,2R)-1-(2,3-dihydro-1,4-benzodioxin-6-yl)-1-hydroxy-3-pyrrolidin-1-ylpropan-2-yl]octanamide
C-NMR spectral analysis
………………..
http://www.google.com/patents/WO2013059119A1?cl=en
http://www.google.com/patents/US7196205
Compound 7
(1R,2R)-Nonanoic acid[2-(2′,3′-dihydro-benzo[1,4]dioxin-6′-yl)-2-hydroxy-1-pyrrolidin-1-ylmethyl-ethyl]-amide
This compound was prepared by the method described for Compound 6 using Nonanoic acid N-hydroxysuccinimide ester. Analytical HPLC showed this material to be 98.4% pure. mp 74–75° C.
1H NMR (CDCl3) δ 6.86–6.76 (m, 3H), 5.83 (d, J=7.3 Hz, 1H), 4.90 (d, J=3.3 Hz, 1H), 4.24 (s, 4H), 4.24–4.18 (m, 1H), 2.85–2.75 (m, 2H), 2.69–2.62 (m, 4H), 2.10 (t, J=7.3 Hz, 2H), 1.55–1.45 (m, 2H), 1.70–1.85 (m, 4H), 1.30–1.15 (m, 10H), 0.87 (t, J=6.9 Hz, 3H) ppm.
Intermediate 4(1R,2R)-2-Amino-1-(2′,3′-dihydro-benzo[1,4]dioxin-6′-yl)-3-pyrrolidin-1-yl-propan-1-ol
Intermediate 3 (5.3 g, 13.3 mmol) was dissolved in methanol (60 mL). Water (6 mL) and trifluoroacetic acid (2.05 m/L, 26.6 mmol, 2 equivalents) were added. After being placed under nitrogen, 20% Palladium hydroxide on carbon (Pearlman’s catalysis, Lancaster or Aldrich, 5.3 g) was added. The mixture was placed in a Parr Pressure Reactor Apparatus with glass insert. The apparatus was placed under nitrogen and then under hydrogen pressure 110–120 psi. The mixture was stirred for 2–3 days at room temperature under hydrogen pressure 100–120 psi. The reaction was placed under nitrogen and filtered through a pad of celite. The celite pad was washed with methanol (100 mL) and water (100 mL). The methanol was removed by rotoevaporation. The aqueous layer was washed with ethyl acetate three times (100, 50, 50 mL). A 10 M NaOH solution (10 mL) was added to the aqueous layer (pH=12–14). The product was extracted from the aqueous layer three times with methylene chloride (100, 100, 50 mL). The combined organic layers were dried with Na2SO4, filtered and rotoevaporated to a colorless oil. The foamy oil was vacuum dried for 2 h. Intermediate 4 was obtained in 90% yield (3.34 g).
Intermediate 3(1R,2R,1″S)-1-(2′,3′-Dihydro-benzo[1,4]dioxin-6′-yl)-2-(2″-hydroxy -1′-phenyl-ethylamino)-3-pyrrolidin-1-yl-propan-1-ol
To a 3-neck flask equipped with a dropping funnel and condenser was added LiAlH4 (Aldrich, 1.2 g, 31.7 mmol, 2.5 equivalents) and anhydrous THF (20 mL) under nitrogen. A solution of Intermediate 2 (5.23 g, 12.68 mmol) in anhydrous THF (75 mL) was added dropwise to the reaction over 15–30 minutes. The reaction was refluxed under nitrogen for 9 hours. The reaction was cooled in an ice bath and a 1M NaOH solution was carefully added dropwise. After stirring at room temperature for 15 minutes, water (50 mL) and ethyl acetate (75 mL) was added. The layers were separated and the aqueous layer was extracted twice with ethyl acetate (75 mL). The combined organic layers were washed with saturated sodium chloride solution (25 mL). After drying with Na2SO4 the solution was filtered and rotoevaporated to yield a colorless to yellow foamy oil. Intermediate 3 was obtained in 99% yield (5.3 g).

|
|
| Systematic (IUPAC) name | |
|---|---|
| N-[(1R,2R)-1-(2,3-Dihydro-1,4-benzodioxin-6-yl)-1-hydroxy-3-(1-pyrrolidinyl)-2-propanyl]octanamide | |
| Clinical data | |
| Trade names | Cerdelga |
|
|
| Identifiers | |
| 491833-29-5 | |
| A16AX10 | |
| PubChem | CID 23652731 |
| ChemSpider | 28475348 |
| ChEBI | CHEBI:82752 |
| Chemical data | |
| Formula | C23H36N2O4 |
| 404.543 g/mol | |
| WO2008150486A2 * | May 30, 2008 | Dec 11, 2008 | Genzyme Corp | 2-acylaminopropoanol-type glucosylceramide synthase inhibitors |
| WO2009045503A1 * | Oct 3, 2008 | Apr 9, 2009 | Genzyme Corp | Method of treating polycystic kidney diseases with ceramide derivatives |
| WO2010014554A1 * | Jul 27, 2009 | Feb 4, 2010 | Genzyme Corporation | Glucosylceramide synthase inhibition for the treatment of collapsing glomerulopathy and other glomerular disease |
| WO2010039256A1 * | Oct 2, 2009 | Apr 8, 2010 | Genzyme Corporation | 2-acylaminopropoanol-type glucosylceramide synthase inhibitors |
………………..
SWEDEN


Alfred Nobel had the unpleasant surprise of reading his own obituary, titled The merchant of death is dead, in a French newspaper.



Nyköping (Sweden)-houses.
Fjallbacka, a colorful fishing Village along the west coast of Sweden
Knights Island, Stockholm, Sweden
 Stockholm, Sweden
Stockholm, Sweden
Sweden Stockholm
Europe Örby Änger – Sweden
 Despite the cold weather, public came and enjoyed different activities. The famous chef, Paul Svensson who works in one of the fanciest and most famous …
Despite the cold weather, public came and enjoyed different activities. The famous chef, Paul Svensson who works in one of the fanciest and most famous …


Vintafolide, EC-145 , MK-8109
mw 1917.041, cas 742092-03-1, mf C86 H109 N21 O26 S2
(2S)-2-[(4-{[(2-amino-4-oxo-3H-pteridin-6-yl)methyl]amino}phenyl)formamido]-4-{[(1S)-1-{[(1S)-4-carbamimidamido-1-{[(1S)-2-carboxy-1-{[(1S)-2-carboxy-1-{[(1R)-1-carboxy-2-({2-[({[(1R,9R,10S,11R,12R,19R)-12-ethyl-4-[(13S,15R,17S)-17-ethyl-17-hydroxy-13-(methoxycarbonyl)-1,11-diazatetracyclo[13.3.1.04,12.05,10]nonadeca-4(12),5,7,9-tetraen-13-yl]-10,11-dihydroxy-5-methoxy-8-methyl-8,16-diazapentacyclo[10.6.1.01,9.02,7.016,19]nonadeca-2,4,6,13-tetraen-10-yl]formohydrazido}carbonyl)oxy]ethyl}disulfanyl)ethyl]carbamoyl}ethyl]carbamoyl}ethyl]carbamoyl}butyl]carbamoyl}-2-carboxyethyl]carbamoyl}butanoic acid
Vincaleukoblastin-23-oic acid, O4-deacetyl-, 2-[(2-mercaptoethoxy)carbonyl]hydrazide, disulfide with N-[4-[[(2-amino-1,4-dihydro-4-oxo-6-pteridinyl)methyl]amino]benzoyl]-L-γ-glutamyl-L-α-aspartyl-L-arginyl-L-α-aspartyl-L-α-aspartyl-L-cysteine
Endocyte innovator
Vintafolide is an investigational targeted cancer therapeutic currently under development by Endocyte and Merck & Co.[1] It is a small molecule drug conjugate consisting of a small molecule targeting the folate receptor, which is overexpressed on certain cancers, such as ovarian cancer, and a potent chemotherapy drug, vinblastine.[2] It is being developed with a companion imaging agent, etarfolatide, that identifies patients that express the folate receptor and thus would likely respond to the treatment with vintafolide.[3] A Phase 3 study evaluating vintafolide for the treatment of platinum-resistant ovarian cancer (PROCEED trial) and a Phase 2b study(TARGET trial) in non-small-cell lung carcinoma (NSCLC) are ongoing.[4] Vintafolide is designed to deliver the toxic vinblastine drug selectively to cells expressing the folate receptor using folate targeting.[5]
A Marketing Authorization Application (MAA) filing for vintafolide and etarfolatide for the treatment of patients withfolate receptor-positive platinum-resistant ovarian cancer in combination with doxorubicin, pegylated liposomal doxorubicin (PLD), has been accepted by the European Medicines Agency.[6] The drug received an orphan drug status in Europe in March 2012.[1] Merck & Co. acquired the development and marketing rights to this experimental cancer drug from Endocyte in April 2012.[1] The drug received orphan drug status in Europe in March 2012.[3]Endocyte remains responsible for the development and commercialization of etarfolatide, a non-invasive companion imaging agent used to identify patients expressing the folate receptor that will likely respond to treatment with vintafolide.[4] Vintafolide is designed to deliver the toxic vinblastine drug selectively to cells expressing the folate receptor using folate targeting.[5]
In 2014 Merck and Endocyte stopped a late-stage study of vintafolide in treating ovarian cancer on the recommendation of a data safety monitoring board, saying that the drug failed to improve progression-free survival.[7]
Vintafolide is folate-conjugated with DAVBLH, which is a derivative of the vinca alkaloid vinblastine.Vinblastine is a microtubule-destabilizing agent that binds tubulin and causes M phase-specific cell cycle arrest and apoptosis of mitotically active cells. Vinblastine is an extremely potent chemotherapeutic agent but has significant toxicities including bone marrow suppression, neurotoxicity, gastrointestinal toxicity and vesicant injury.
Endocyte’s desacetylvinblastinehydrazide/folate conjugate (EC-145) is a folate-targeted cytotoxic anticancer drug in early development for the treatment of non-small cell lung cancer (NSCLC) and breast cancer. The compound had been pre-registered in the E.U. by Merck for the treatment of ovarian cancer, but the application was withdrawn due to lack of efficacy.
In 2012, the product was licensed to Merck & Co. by Endocyte for worldwide exclusive development and commercialization. In 2014, however, this license agreement was terminated and Endocyte regained all rights.
Folates can serve as one-carbon donors in reactions that are critical in the de novo biosynthesis of purines and thymidylate, amino acid metabolism and methylation reactions. Folate can enter a cell by two routes: RFC or by membrane-bound FRs. RFC is a bidirectional anion transporter that is the normal entry method for reduced folates in most cells. By contrast, FRs are expressed in a limited distribution in normal tissues but are overexpressed in multiple cancers including ovarian, lung, breast and colorectal cancer. FRs bind folate derivatives with high affinity and mediate their internalization by endocytosis. Given that FRs are not typically expressed on the luminal surface of epithelial cells, making them inaccessible to normal circulation, they are attractive therapeutic targets with limited toxicity. In addition to the therapeutic agent vintafolide, a radiodiagnostic agent (99mTc-etarfolatide [EC20]) has been developed to allow single-photon emission computed tomography (SPECT) imaging to identify FR-expressing tissues (tumors).
In 2012, orphan drug designations were assigned in the E.U. for the treatment of ovarian cancer and to be used with folic acid for the diagnosis of positive folate-receptor status in ovarian cancer. In 2013, orphan drug designation was assigned in the U.S. for the treatment of ovarian cancer.
Vintafolide is a water-soluble derivative of folic acid and the vinca alkaloid DAVLBH. The molecules are connected through a hydrophilic L-peptide spacer and a disulfide linker (Figure 1). The disulfide linker serves as a cleavable bond that is necessary for drug release following receptor mediated endocytosis. The disulfide bond is reduced in the acidic environment of the endosome, leading to efficient release of vinblastine.
Vintafolide.
DAVBLH: Desacetylvinblastine hydrazide
Structure of vintafolide and mechanism of release of the payload in the endosome.
Folate is required for cell division, and rapidly dividing cancer cells often express folate receptors in order to capture enough folate to support rapid cell growth. Elevated expression of the folate receptor occurs in many diseases, including other aggressively growing cancers and inflammatory disorders.[8] Vintafolide binds to the folate receptor and is subsequently taken up by the cell through a natural internalization process called endocytosis. Once inside the cell, vintafolide’s linker releases the chemotherapy drug which kills the cell.[3]
……………
Bioorganic & Medicinal Chemistry Letters (2006), 16(19), 5093-5096
http://www.sciencedirect.com/science/article/pii/S0960894X06008079
An efficient synthesis of the folate receptor (FR) targeting conjugate EC145 is described. EC145 is a water soluble derivative of the vitamin folic acid and the potent cytotoxic agent, desacetylvinblastine monohydrazide. Both molecules are connected in regioselective manner via a hydrophilic peptide spacer and a reductively labile disulfide linker.

………approach for the design and regioselective synthesis of a FA-vinca alkaloid conjugate 1 (EC145,BELOW). As indicated in the retrosynthetic scheme, 1 can be assembled by tethering a FA-Spacer unit 2 to the highly potent cytotoxic molecule, desacetylvinblastine monohydrazide 3, via a linker containing a reducible disulfide bond. The latter is important for drug delivery applications since real-time imaging using a fluorescence resonance energy transfer technique has recently demonstrated that reduction-mediated release of the drug cargo from a disulfide linked FA-conjugate efficiently occurs within the endosomes of cancer cells.
Scheme 1.
Reagents and conditions: (i) a—Fmoc-Asp(OtBu)-OH, PyBOP, DIPEA, RT, 1 h; b—20% piperidine/DMF, rt, 10 min; (ii) a—Fmoc-Arg(Pbf)-OH, PyBOP, DIPEA, rt, 1 h; b—20% piperidine/DMF, rt, 10 min; (iii) a—Fmoc-Glu-OtBu, PyBOP, DIPEA, rt, 1 h; b—20% piperidine/DMF, rt, 10 min; (iv) N10-TFA-pteroic acid, PyBOP, DIPEA, rt, 1.5 h; (v) TFA/H2O/TIPS/EDT (92.5:2.5:2.5:2.5), rt, 1 h; (vi) aq NH4OH, pH 9.3, rt, 1 h.
EC145 (D2O, 300 MHz): δ 8.67 (s, 1H, FA H-7), 7.50 (br s, 1H, VLB H-11′), 7.30–7.40 (br s, 1H, VLB H-14′), 7.35 (d, 2H, J = 7.8 Hz, FA H-12 & 16), 7.25 (m, 1H, VLB H-13′), 7.05 (br s, 1H, VLB H-12′), 6.51 (d, 2H, J = 8.7 Hz, FA H-13 & 15), 6.4 (s, 2H, VLB H-14 & 17), 5.65 (m, 1H, VLB H-7), 5.5 (m, 1H, VLB H-6), 4.15 (m,1H, VLB H-8′), 3.82 (s, 3H, VLB C18′ –CO2CH3), 3.69 (s, 3H, VLB C16 –OCH3), 2.8 (s, 3H, VLB N–CH3), 1.35 (br s, 1H, VLB H-3′), 1.15 (m, 1H, VLB H-2′), 0.9 (t, 3H, J = 7 Hz, VLB H-21′), 0.55 (t, 3H, J = 6.9 Hz, VLB H-21).
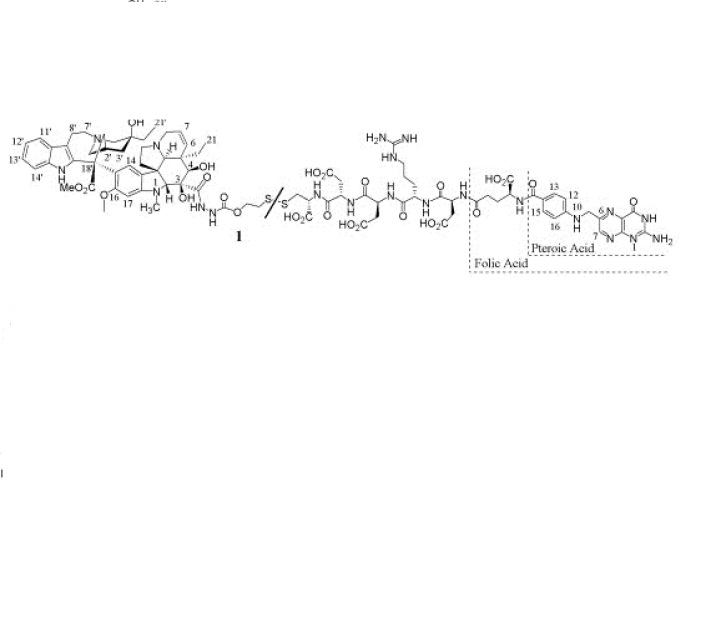 CLICK ON IMAGE FOR CLEAR VIEW
CLICK ON IMAGE FOR CLEAR VIEW
…………
WO 2004069159
http://www.google.com/patents/WO2004069159A2?cl=en
EXAMPLE 16b
The compounds of Examples 16a and 16b were prepared from the peptidyl fragment Pte-Glu-Asp-Arg-Asp-Asp-Cys-OH , prepared according to the general procedure described in Scheme 12. The Michael addition of this peptidyl fragment to the maleimido derivative of seco-CBI-bis-indole resulted in the folate conjugates Example 16a. The peptidyl fragment also reacted with either the thiosulfonate or pyridyldithio-activated vinblastine to form Example 16b. The maleimido derivative of seco-CBI-bis-indole, and the thiosulfonate and pyridyldithio- activated vinblastine intermediates were prepared using the procedures described herein for other examples.
……………..
https://www.google.com/patents/WO2012142281A1?cl=en
Folate-targeted drugs have been developed and are being tested in clinical trials as cancer therapeutics. EC145, also known as vintafolide, comprises a highly potent vinca alkaloid cytotoxic compound, desacetylvinblastine hydrazide (DAVLBH), conjugated to folate. The EC 145 molecule targets the folate receptor found at high levels on the surface of epithelial tumors, including non-small cell lung carcinomas (NSCLC), ovarian, endometrial and renal cancers, and others, including fallopian tube and primary peritoneal carcinomas. It is believed that EC 145 binds to tumors that express the folate receptor delivering the vinca moiety directly to cancer cells while avoiding normal tissue. Thus, upon binding, EC 145 enters the cancer cell via endocytosis, releases DAVLBH and causes cell death or inhibits cell function. EC 145 has the following formula
EC145
and has been accorded the Chemical Abstracts Registry Number 742092-03-1. As used herein, according to the context, the term EC 145 means the compound, or a pharmaceutically acceptable salt thereof; and the compound may be present in a solid, solution or suspension in an ionized form, including a protonated form. EC145 is disclosed in U.S. Patent No. 7,601,332; and particular uses and an aqueous liquid pH 7.4, phosphate-buffered formulation for intravenous administration are disclosed in WO 2011/014821. As described in WO 2011/014821, it is necessary to store the aqueous liquid formulation in the frozen state to ensure its stability. To avoid this necessity, a formulation is needed which has adequate stability at ambient temperature.
As one aspect of the invention described herein, there is provided a pharmaceutical composition of EC145 which is a lyophilized solid which has adequate stability for storage at ambient temperature and which is capable of redissolving in an aqueous diluent prior to administration.
In another aspect of the invention, there is provided a pharmaceutical composition of EC 145 which is an X-ray amorphous solid which has adequate stability for storage at ambient temperature and which is capable of redissolving in an aqueous diluent prior to administration.
| Systematic (IUPAC) name | |
|---|---|
|
N-(4-{[(2-Amino-4-oxo-1,4-dihydropteridin-6-yl)methyl]amino}benzoyl)-L-γ-glutamyl-L-α-aspartyl-L-arginyl-L-α-aspartyl-L-α-aspartyl-L-cysteine disulfide with methyl (5S,7R,9S)-5-ethyl-9-[(3aR,4R,5S,5aR,10bR,13aR)-3a-ethyl-4,5-dihydroxy-8-methoxy-6-methyl-5-({2-[(2-sulfanylethoxy)carbonyl]hydrazinyl}carbonyl)-3a,4,5,5a,6,11,12,13a-octahydro-1H-indolizino[8,1-cd]carbazol-9-yl]-5-hydroxy-1,4,5,6,7,8,9,10-octahydro-2H-3,7-methanoazacycloundecino[5,4-b]indol-9-carboxylate
|
|
| Clinical data | |
| Legal status |
|
| Identifiers | |
| CAS Registry Number | 742092-03-1 |
| ATC code | L01CA06 |
| ChemSpider | 27444385 |
| Synonyms | EC-145 |
| Chemical data | |
| Formula | C86H109N21O26S2 |
| Molecular mass | 1917 g/mol |
| WO2008098970A1 * | Feb 13, 2008 | Aug 21, 2008 | Pf Medicament | Anhydrous crystalline vinflunine salts, method of preparation and use thereof as a drug and means of vinflunine purification |
| WO2010150100A1 * | Jun 23, 2010 | Dec 29, 2010 | Entarco Sa | The use of spinosyns and spinosyn compositions against diseases caused by protozoans, viral infections and cancer |
| WO2011014821A1 * | Jul 30, 2010 | Feb 3, 2011 | Endocyte, Inc. | Folate-targeted diagnostics and treatment |
| US20100247669 * | Sep 30, 2010 | Cerulean Pharma Inc. | Polymer-agent conjugates, particles, compositions, and related methods of use |
////////Vintafolide, BMS-753493, DAVBLH, Desacetylvinblastine hydrazide, EC-145 , MK-8109 , phase 2

Ombitasvir; ABT-267; ABT 267; UNII-2302768XJ8; 1258226-87-7;
| C50H67N7O8 | |
| Molecular Weight: | 894.10908 g/mol |
|---|
Anti-Viral Compounds [US2010317568]
methyl N-[(2S)-1-[(2S)-2-[[4-[(2S,5S)-1-(4-tert-butylphenyl)-5-[4-[[(2S)-1-[(2S)-2-(methoxycarbonylamino)-3-methylbutanoyl]pyrrolidine-2-carbonyl]amino]phenyl]pyrrolidin-2-yl]phenyl]carbamoyl]pyrrolidin-1-yl]-3-methyl-1-oxobutan-2-yl]carbamate
Ombitasvir is an antiviral drug for the treatment of hepatitis C virus (HCV) infection. In the United States, it is approved by theFood and Drug Administration for use in combination with paritaprevir, ritonavir and dasabuvir in the product Viekira Pak for the treatment of HCV genotype 1,[1][2] and with paritaprevir and ritonavir in the product Technivie for the treatment of HCV genotype 4.[3][4]
Ombitasvir is in phase II clinical development at AbbVie for the treatment of chronic hepatitis C infection in combination with ABT-450/ritonavir and, in combination with peginterferon alpha-2a/ribavirin (pegIFN/RBV) in treatment naïve Hepatitis C virus (HCV) genotype 1 infected patients.
Ombitasvir is part of a fixed-dose formulation with ABT-450/ritonavir that is approved in the U.S. and the E.U.
Ombitasvir acts by inhibiting the HCV protein NS5A.[5]
In 2013, breakthrough therapy designation was assigned in the U.S. for the treatment of genotype 1 hepatitis C in combination with ABT-450, ritonavir and ABT-333, with and without ribavirin.

http://pubs.acs.org/doi/suppl/10.1021/jm401398x/suppl_file/jm401398x_si_001.pdf
We describe here N-phenylpyrrolidine-based inhibitors of HCV NS5A with excellent potency, metabolic stability, and pharmacokinetics. Compounds with 2S,5S stereochemistry at the pyrrolidine ring provided improved genotype 1 (GT1) potency compared to the 2R,5Ranalogues. Furthermore, the attachment of substituents at the 4-position of the central N-phenyl group resulted in compounds with improved potency. Substitution with tert-butyl, as in compound 38 (ABT-267), provided compounds with low-picomolar EC50 values and superior pharmacokinetics. It was discovered that compound 38 was a pan-genotypic HCV inhibitor, with an EC50 range of 1.7–19.3 pM against GT1a, -1b, -2a, -2b, -3a, -4a, and -5a and 366 pM against GT6a. Compound 38 decreased HCV RNA up to 3.10 log10 IU/mL during 3-day monotherapy in treatment-naive HCV GT1-infected subjects and is currently in phase 3 clinical trials in combination with an NS3 protease inhibitor with ritonavir (r) (ABT-450/r) and an NS5B non-nucleoside polymerase inhibitor (ABT-333), with and without ribavirin.
PATENT
WO 2011156578
dimethyl (2S,2,S)-l,l ‘-((2S,2’S)-2,2′-(4,4’-((2S,5S)-l-(4-fert-butylphenyl)pyrrolidine- 2,5-diyl)bis(4, 1 -phenylene))bis(azanediyl)bis(oxomethylene)bis(pyrrolidine-2, 1 -diyl))bis(3- methyl- l-oxobutane-2,l-diyl)dicarbamate
hereinafter Compound IA),..http://www.google.com/patents/WO2011156578A1?cl=en
……………………………..
PATENT
US 20100317568
https://www.google.co.in/patents/US20100317568
Example 34
Dimethyl (2S,2’S)-l,r-((2S,2’S)-2,2′-(4,4′-((2S,5S)-1-(4-tert-butylphenyl)pyrrolidine-2,5-diyl)bis(4,l- phenylene))bis(azanediyl)bis(oxomethylene)bis(pyrrolidine-2, 1 -diyl))bis(3-methyl- 1 -oxobutane-2, 1 – diyl)dicarbamate and
Dimethyl (2S,2’S)-l,r-((2S,2’S)-2,2′-(4,4′-((2R,5R)-1-(4-ter/’-butylphenyl)pyrrolidine-2,5- diyl)bis(4, 1 -phenylene))bis(azanediyl)bis(oxomethylene)bis(pyrrolidine-2, 1 -diyl))bis(3-methyl- 1 – oxobutane-2, 1 -diyl)dicarbamate
Example 34A l-(4-fer?-butylphenyl)-2,5-bis(4-nitrophenyl)pyrrolidine The product from Example 1C (3.67 g, 7.51 mmol) and 4-tert-butylaniline (11.86 ml, 75 mmol) in DMF (40 ml) was stirred under nitrogen at 50 °C for 4 h. The resulting mixture was diluted into ethyl acetate, treated with IM HCl, stirred for 10 minutes and filtered to remove solids. The filtrate organic layer was washed twice with brine, dried with sodium sulfate, filtered and evaporated. The residue was purified by chromatography on silica gel eluting with ethyl acetate in hexane (5% to 30%) to give a solid. The solid was triturated in a minimal volume of 1 :9 ethyl acetate/hexane to give a light yellow solid as a mixture of trans and cis isomers (1.21 g, 36%).
Example 34B 4,4′-((2S,5S)-1-(4-tert-butylphenyl)pyrrolidine-2,5-diyl)dianiline and 4,4′-((2R,5R)-1-(4-fert- butylphenyl)pyrrolidine-2,5-diyl)dianiline To a solution of the product from Example 34A (1.1 g, 2.47 mmol) in ethanol (20 ml) and
THF (20 ml) was added PtC>2 (0.22 g, 0.97 mmol) in a 50 ml pressure bottle and stirred under 30 psi hydrogen at room temperature for 1 h. The mixture was filtered through a nylon membrane and evaporated. The residue was purified by chromatography on silica gel eluting with ethyl acetate in hexane (20% to 60%). The title compound eluted as the first of 2 stereoisomers (trans isomer, 0.51 g, 54%).
Example 34C
(2S,2’S)-tert-Butyl 2,2′-(4,4′-((2S,5S)-1-(4-fer/’-butylphenyl)pyrrolidine-2,5-diyl)bis(4,l- phenylene))bis(azanediyl)bis(oxomethylene)dipyrrolidine- 1 -carboxylate and (2S,2’S)-tert-Butyl 2,2′- (4,4′-((2R,5R)-1-(4-tert-butylphenyl)pyrrolidine-2,5-diyl)bis(4,l- phenylene))bis(azanediyl)bis(oxomethylene)dipyrrolidine-1-carboxylate To a mixture of the product from Example 34B (250 mg, 0.648 mmol), (S)-1-(tert- butoxycarbonyl)pyrrolidine-2-carboxylic acid (307 mg, 1.427 mmol) and HATU (542 mg, 1.427 mmol) in DMSO (10 ml) was added Hunig’s base (0.453 ml, 2.59 mmol). The reaction mixture was stirred at room temperature for 1 h. The mixture was partitioned with ethyl acetate and water. The organic layer was washed with brine, dried with sodium sulfate, filtered and evaporated. The residue was purified by chromatography on silica gel eluting with ethyl acetate in hexane (10% to 50%) to give the title compound (500 mg, 99%).
Example 34D
(2S,2’S)-N,N’-(4,4′-((2S,5S)-1-(4-ter/’-butylphenyl)pyrrolidine-2,5-diyl)bis(4,l- phenylene))dipyrrolidine-2-carboxamide and (2S,2’S)-N,N’-(4,4′-((2R,5R)-1-(4-tert- butylphenyl)pyrrolidine-2,5-diyl)bis(4,l-phenylene))dipyrrolidine-2-carboxamide To the product from Example 34C (498 mg, 0.638 mmol) in dichloromethane (4 ml) was added TFA (6 ml). The reaction mixture was stirred at room temperature for 1 h and concentrated in vacuo. The residue was partitioned between 3: 1 CHCl3dsopropyl alcohol and saturated aq. NaHCO3. The aqueous layer was extracted by 3: 1 CHCl3:isopropyl alcohol again. The combined organic layers were dried over
filtered and concentrated to give the title compound (345 mg, 93%).
Example 34E Dimethyl (2S,2’S)-l,r-((2S,2’S)-2,2′-(4,4′-((2S,5S)-1-(4-fert-butylphenyl)pyrrolidine-2,5-diyl)bis(4,l- phenylene))bis(azanediyl)bis(oxomethylene)bis(pyrrolidine-2, 1 -diyl))bis(3-methyl- 1 -oxobutane-2, 1 – diyl)dicarbamate and
Dimethyl (2S,2’S)-1, r-((2S,2’S)-2,2′-(4,4′-((2R,5R)-1-(4-fert-butylphenyl)pyrrolidine-2,5- diyl)bis(4, 1 -phenylene))bis(azanediyl)bis(oxomethylene)bis(pyrrolidine-2, 1 -diyl))bis(3-methyl- 1 – oxobutane-2, 1 -diyl)dicarbamate
The product from Example 34D (29.0 mg, 0.050 mmol), (S)-2-(methoxycarbonylamino)-3- methylbutanoic acid (19.27 mg, 0.110 mmol), EDAC (21.09 mg, 0.110 mmol), HOBT (16.85 mg,
0.110 mmol) and N-methylmorpholine (0.027 ml, 0.250 mmol) were combined in DMF (2 ml). The reaction mixture was stirred at room temperature for 3 h. The mixture was partitioned with ethyl acetate and water. The organic layer was washed with brine twice, dried with sodium sulfate, filtered and evaporated. The residue was purified by chromatography on silica gel eluting with ethyl acetate in hexane (50% to 80%) to give a solid. The solid was triturated with ethyl acetate/hexane to give the title compound (13 mg, 29%). 1H NMR (400 MHz, DMSO-D6) δ ppm 0.85 – 0.95 (m, 12 H) 1.11 (s, 9 H) 1.59 – 1.65 (m, 2 H) 1.79 – 2.04 (m, 8 H) 2.10 – 2.18 (m, 2 H) 2.41-2.46 (m, 2H) 3.52 (s, 6 H)
3.57 – 3.67 (m, 2 H) 3.76 – 3.86 (m, 2 H) 4.00 (t, J=7.56 Hz, 2 H) 4.39 – 4.46 (m, 2 H) 5.15 (d, J=7.00
Hz, 2 H) 6.17 (d, J=7.70 Hz, 2 H) 6.94 (d, J=8.78 Hz, 2 H) 7.13 (d, J=7.37 Hz, 4 H) 7.30 (d, J=8.20
Hz, 2 H) 7.50 (d, J=8.24 Hz, 4 H) 9.98 (s, 2 H); (ESI+) m/z 895 (M+H)+. The title compound showed an EC50 value of less than about 0.1 nM in HCV lb-Conl replicon assays in the presence of 5% FBS.
Example 35
Dimethyl (2S,2’S)-l,r-((2S,2’S)-2,2′-(4,4′-((2S,5S)-1-(4-tert-butylphenyl)pyrrolidine-2,5-diyl)bis(4,l- phenylene))bis(azanediyl)bis(oxomethylene)bis(pyrrolidine-2, 1 -diyl))bis(3-methyl- 1 -oxobutane-2, 1 – diyl)dicarbamate
The product from Example 34E was purified by chiral chromatography on a Chiralpak AD-H semi-prep column eluting with a 2:1 mixture of hexane:(2: l isopropyl alcohol: EtOH). The title compound was the first of the 2 diastereomers to elute. 1H NMR (400 MHz, DMSO-D6) δ ppm 0.88 (d, J=6.61 Hz, 6 H) 0.93 (d, J=6.72 Hz, 6 H) 1.11 (s, 9 H) 1.63 (d, J=5.42 Hz, 2 H) 1.80 – 2.04 (m, 8 H) 2.09 – 2.19 (m, 2 H) 2.44 – 2.47 (m, 2 H) 3.52 (s, 6 H) 3.59 – 3.66 (m, 2 H) 3.77 – 3.84 (m, 2 H) 4.02 (t, J=8.40 Hz, 2 H) 4.42 (dd, J=7.86, 4.83 Hz, 2 H) 5.14 (d, J=6.18 Hz, 2 H) 6.17 (d, J=8.67 Hz, 2 H) 6.94 (d, J=8.78 Hz, 2 H) 7.13 (d, J=8.46 Hz, 4 H) 7.31 (d, J=8.35 Hz, 2 H) 7.50 (d, J=8.35 Hz, 4 H) 9.98 (s, 2 H). The title compound showed an EC50 value of less than about 0.1 nM in HCV Ib- Conl replicon assays in the presence of 5% FBS.
Example 36 Dimethyl (2S,2’S)-1, r-((2S,2’S)-2,2′-(4,4′-((2R,5R)-1-(4-fert-butylphenyl)pyrrolidine-2,5- diyl)bis(4, 1 -phenylene))bis(azanediyl)bis(oxomethylene)bis(pyrrolidine-2, 1 -diyl))bis(3-methyl- 1 – oxobutane-2, 1 -diyl)dicarbamate
The product from Example 34E was purified by chiral chromatography on a Chiralpak AD-H semi-prep column eluting with a 2:1 mixture of hexane:(2: l isopropyl alcohol: EtOH). The title compound was the second of 2 diastereomers to elute. 1H NMR (400 MHz, DMSO-D6) δ ppm 0.87
(d, J=6.51 Hz, 6 H) 0.92 (d, J=6.72 Hz, 6 H) 1.11 (s, 9 H) 1.63 (d, J=5.53 Hz, 2 H) 1.82 – 2.04 (m, 8
H) 2.09-2.18 (m, 2 H) 2.41 – 2.47 (m, 2 H) 3.52 (s, 6 H) 3.58 – 3.67 (m, 2 H) 3.75 – 3.84 (m, 2 H) 4.02
(t, J=7.26 Hz, 2 H) 4.43 (dd, J=7.92, 4.88 Hz, 2 H) 5.14 (d, J=6.18 Hz, 2 H) 6.17 (d, J=8.78 Hz, 2 H) 6.94 (d, J=8.67 Hz, 2 H) 7.12 (d, J=8.46 Hz, 4 H) 7.31 (d, J=8.35 Hz, 2 H) 7.49 (d, J=8.46 Hz, 4 H)
9.98 (s, 2 H). The title compound showed an EC50 value of less than about 0.1 nM in HCV lb-Conl replicon assays in the presence of 5% FBS.
Example 37 Dimethyl (2S,2’S)-l,r-((2S,2’S)-2,2′-(4,4′-((2S,5S)-1-(4-fert-butylphenyl)pyrrolidine-2,5-diyl)bis(4,l- phenylene))bis(azanediyl)bis(oxomethylene)bis(pyrrolidine-2, 1 -diyl))bis(3-methyl- 1 -oxobutane-2, 1 – diyl)dicarbamate
Example 37A (S)-2,5-dioxopyrrolidin-1-yl 2-(methoxycarbonylamino)-3-methylbutanoate To a mixture of (S)-2-(methoxycarbonylamino)-3-methylbutanoic acid (19.66 g, 112 mmol) and N-hydroxysuccinimide (13.29g, 116 mmol) was added ethyl acetate (250 ml), and the mixture was cooled to 0-5 °C. Diisopropylcarbodiimide (13.88 g, 110 mmol) was added and the reaction mixture was stirred at 0-5 °C for about 1 hour. The reaction mixture was warmed to room temperature. The solids (diisopropylurea by-product) were filtered and rinsed with ethyl acetate. The filtrate was concentrated in vacuo to an oil. Isopropyl alcohol (200 ml) was added to the oil and the mixture was heated to about 50 °C to obtain a homogeneous solution. Upon cooling, crystalline solids formed. The solids were filtered and washed with isopropyl alcohol (3 x 20 ml) and dried to give the title compound as a white solid (23.2 g, 77% yield).
Example 37B
(S)- 1 -((S)-2-(methoxycarbonylamino)-3-methylbutanoyl)pyrrolidine-2-carboxylic acid To a mixture of L-proline (4.44g, 38.6 mmol), water (20 ml), acetonitrile (20 ml) and DIEA (9.5 g, 73.5 mmol) was added a solution of the product from Example 37A (1Og, 36.7 mmol) in acetonitrile (20 inL) over 10 minutes. The reaction mixture was stirred overnight at room temperature. The solution was concentrated under vacuum to remove the acetonitrile. To the resulting clear water solution was added 6N HCl (9 ml) until pH ~ 2 .The solution was transferred to a separatory funnel and 25% NaCl (10 ml) was added and the mixture was extracted with ethyl acetate (75 ml), and then again with ethyl acetate (6 x 20 ml), and the combined extracts were washed with 25% NaCl (2 x 10ml). The solvent was evaporated to give a thick oil. Heptane was added and the solvent was evaporated to give a foam, which was dried under high vacuum. Diethyl ether was added and the solvent was evaporated to give a foam, which was dried under high vacuum to give the title compound (10.67g) as a white solid.
The compound of Example 37B can also be prepreared according to the following procedure: To a flask was charged L- valine (35 g, 299 mmol), IN sodium hydroxide solution (526 ml,
526 mmol) and sodium carbonate (17.42 g, 164 mmol). The mixture was stirred for 15 min to dissolve solids and then cooled to 15 °C. Methyl chloroformate (29.6 g, 314 mmol) was added slowly to the reaction mixture. The mixture was then stirred at rt for 30 min. The mixture was cooled to 15 °C and pH adjusted to -5.0 with concentrated HCl solution. 100 inL of 2-methytetrahydrofuran (2- MeTHF) was added and the adjustment of pH continued until the pH reached ~ 2.0. 150 mL of 2- MeTHF was added and the mixture was stirred for 15 min. Layers were separated and the aqueous layer extracted with 100 mL of 2-MeTHF. The combined organic layer was dried over anhyd Na2SC^ and filtered, and Na2SC^ cake was washed with 50 mL of 2-MeTHF. The product solution was concentrated to ~ 100 mL, chased with 120 mL of IPAc twice. 250 mL of heptanes was charged slowly and then the volume of the mixture was concentrated to 300 mL. The mixture was heated to 45 °C and 160 mL of heptanes charged. The mixture was cooled to rt in 2h, stirred for 30 min, filtered and washed with 2-MeTHF/heptanes mixture (1:7, 80 inL). The wetcake was dried at 55 °C for 24 h to give 47.1 g of Moc-L- VaI-OH product as a white solid (90%).
Moc-L- VaI-OH (15O g, 856 mmol), HOBt hydrate (138 g, 899 mmol) and DMF (1500 ml) were charged to a flask. The mixture was stirred for 15 min to give a clear solution. EDC hydrochloride (172 g, 899 mmol) was charged and mixed for 20 min. The mixture was cooled to 13
°C and (L)-proline benzyl ester hydrochloride (207 g, 856 mmol) charged. Triethylamine (109 g,
1079 mmol) was then charged in 30 min. The resulting suspension was mixed at rt for 1.5 h. The reaction mixture was cooled to 15 °C and 1500 mL of 6.7% NaHCO3 charged in 1.5 h, followed by the addition of 1200 mL of water over 60 min. The mixture was stirred at rt for 30 min, filtered and washed with water/DMF mixture (1 :2, 250 mL) and then with water (1500 mL). The wetcake was dried at 55 °C for 24 h to give 282 g of product as a white solid (90%).
The resulting solids (40 g) and 5% Pd/ Alumina were charged to a Parr reactor followed by THF (160 mL). The reactor was sealed and purged with nitrogen (6 x 20 psig) followed by a hydrogen purge (6 x 30 psig). The reactor was pressurized to 30 psig with hydrogen and agitated at room temperature for approximately 15 hours. The resulting slurry was filtered through a GF/F filter and concentrated to approximately 135 g solution. Heptane was added (120 mL), and the solution was stirred until solids formed. After an addition 2 – 3 hours additional heptane was added drop-wise (240 mL), the slurry was stirred for approximately 1 hour, then filtered. The solids were dried to afford the title compound.
Example 37C
(lR,4R)-1,4-bis(4-nitrophenyl)butane-1,4-diyl dimethanesulfonate
The product from Example 32 (5.01 g, 13.39 mmol) was combined with 2- methyltetrahydrofuran (70 mL) and cooled to -5 °C, and N,N-diisopropylethylamine (6.81 g, 52.7 mmol) was added over 30 seconds. Separately, a solution of methanesulfonic anhydride (6.01 g, 34.5 mmol) in 2-methyltetrahydrofuran (30 mL) was prepared and added to the diol slurry over 3 min., maintaining the internal temperature between -15 °C and -25 °C. After mixing for 5 min at -15 °C, the cooling bath was removed and the reaction was allowed to warm slowly to 23 °C and mixed for 30 minutes. After reaction completion, the crude slurry was carried immediately into the next step.
Example 37D
(2S,5S)-1-(4-tert-butylphenyl)-2,5-bis(4-nitrophenyl)pyrrolidine
To the crude product solution from Example 37C (7.35 g, 13.39 mmol) was added 4-tert- butylaniline (13.4 g, 90 mmol) at 23 °C over 1 minute. The reaction was heated to 65 °C for 2 h. After completion, the reaction mixture was cooled to 23 °C and diluted with 2-methyltetrahydrofuran (100 mL) and 1 M HCl (150 mL). After partitioning the phases, the organic phase was treated with 1 M HCl (140 mL), 2-methyltetrahydrofuran (50 mL), and 25 wt% aq. NaCl (100 mL), and the phases were partitioned. The organic phase was washed with 25 wt% aq. NaCl (50 mL), dried over MgSO/t, filtered, and concentrated in vacuo to approximately 20 mL. Heptane (30 mL) and additional 2- methyltetrahydrofuran were added in order to induce crystallization. The slurry was concentrated further, and additional heptane (40 mL) was slowly added and the slurry was filtered, washing with 2- methyltetrahydrofuran:heptane (1:4, 20 mL). The solids were suspended in MeOH (46 mL) for 3 h, filtered, and the wet solid was washed with additional MeOH (18 mL). The solid was dried at 45 °C in a vacuum oven for 16 h to provide the title compound (3.08 g, 51% 2-step yield).
Example 37E
4,4′-((2S,5S)-1-(4-tert-butylphenyl)pyrrolidine-2,5-diyl)dianiline
To a 160 ml Parr stirrer hydrogenation vessel was added the product from Example 37D (2 g, 4.49 mmol), followed by 60 ml of THF, and Raney Nickel Grace 2800 (1 g, 50 wt% (dry basis)) under a stream of nitrogen. The reactor was assembled and purged with nitrogen (8 x 20 psig) followed by purging with hydrogen (8 x 30 psig). The reactor was then pressurized to 30 psig with hydrogen and agitation (700 rpm) began and continued for a total of 16 h at room temperature. The slurry was filtered by vacuum filtration using a GF/F Whatman glass fiber filter. Evaporation of the filtrate to afford a slurry followed by the addition heptane and filtration gave the crude title compound, which was dried and used directly in the next step.
Example 37F dimethyl (2S,2’S)-l,r-((2S,2’S)-2,2′-(4,4′-((2S,5S)-1-(4-tert-butylphenyl)pyrrolidine-2,5-diyl)bis(4, l- phenylene)bis(azanediyl)bis(oxomethylene))bis(pyrrolidine-2, 1 -diyl))bis(3-methyl- 1 -oxobutane-2, 1 – diy 1) die arb amate To a solution of the product from Example 37E (1.64 g, 4.25 mmol) in DMF (20 ml), the product from Example 37B (2.89 g, 10.63 mmol), and HATU (4.04 g, 10.63 mmol) in DMF (15OmL) was added triethylamine (1.07 g, 10.63 mmol), and the solution was stirred at room temperature for 90 min. To the reaction mixture was poured 20 mL of water, and the white precipitate obtained was filtered, and the solid was washed with water (3×5 mL). The solid was blow dried for Ih. The crude material was loaded on a silica gel column and eluted with a gradient starting with ethyl acetate/ heptane (3/7), and ending with pure ethyl acetate. The desired fractions were combined and solvent distilled off to give a very light yellow solid, which was dried at 45 °C in a vacuum oven with nitrogen purge for 15 h to give the title compound (2.3 g, 61% yield). 1H NMR (400 MHz, DMSO- D6) δ ppm 0.88 (d, J=6.61 Hz, 6 H) 0.93 (d, J=6.72 Hz, 6 H) 1.11 (s, 9 H) 1.63 (d, J=5.42 Hz, 2 H) 1.80 – 2.04 (m, 8 H) 2.09 – 2.19 (m, 2 H) 2.44 – 2.47 (m, 2 H) 3.52 (s, 6 H) 3.59 – 3.66 (m, 2 H) 3.77 – 3.84 (m, 2 H) 4.02 (t, J=8.40 Hz, 2 H) 4.42 (dd, J=7.86, 4.83 Hz, 2 H) 5.14 (d, J=6.18 Hz, 2 H) 6.17 (d, J=8.67 Hz, 2 H) 6.94 (d, J=8.78 Hz, 2 H) 7.13 (d, J=8.46 Hz, 4 H) 7.31 (d, J=8.35 Hz, 2 H) 7.50 (d, J=8.35 Hz, 4 H) 9.98 (s, 2 H).
Alternately, the product from example 37E (11.7 g, 85 wt%, 25.8 mmol) and the product from example 37B (15.45 g, 56.7 mmol) are suspended in EtOAc (117 mL), diisopropylethylamine (18.67 g, 144 mmol) is added and the solution is cooled to 0 °C. In a separate flask, 1-propanephosphonic acid cyclic anhydride (T3P®) (46.0 g, 50 wt% in EtOAc, 72.2 mmol) was dissolved in EtOAc (58.5 mL), and charged to an addition funnel. The T3P solution is added to the reaction mixture drop-wise over 3-4 h and stirred until the reaction is complete. The reaction is warmed to room temperature,and washed with IM HCl/7.5 wt% NaCl (100 mL), then washed with 5% NaHCO3 (100 mL), then washed with 5% NaCl solution (100 mL). The solution was concentrated to approximately 60 mL, EtOH (300 mL) was added, and the solution was concentrated to 84 g solution.
A portion of the EtOH solution of product (29 g) was heated to 40 °C, and added 134 g 40 w% EtOH in H2O. A slurry of seeds in 58 wt/wt% EtOH/H2O was added, allowed to stir at 40 °C for several hours, then cooled to 0 °C. The slurry is then filtered, and washed with 58wt/wt% EtOH/H2O. The product is dried at 40 – 60 °C under vacuum, and then rehydrated by placing a tray of water in the vacuum oven to give the title compound. The title compound showed an EC50 value of less than about 0.1 nM in HCV lb-Conl replicon assays in the presence of 5% FBS.
……………..
PATENT
http://www.google.com/patents/EP2337781A2?cl=en
Example 34
Dimethyl (2S,2’S)-l,r-((2S,2’S)-2,2′-(4,4′-((2S,5S)-1-(4-tert-butylphenyl)pyrrolidine-2,5-diyl)bis(4,l- phenylene))bis(azanediyl)bis(oxomethylene)bis(pyrrolidine-2, 1 -diyl))bis(3-methyl- 1 -oxobutane-2, 1 – diyl)dicarbamate and
Dimethyl (2S,2’S)-l,r-((2S,2’S)-2,2′-(4,4′-((2R,5R)-1-(4-ter/’-butylphenyl)pyrrolidine-2,5- diyl)bis(4, 1 -phenylene))bis(azanediyl)bis(oxomethylene)bis(pyrrolidine-2, 1 -diyl))bis(3-methyl- 1 – oxobutane-2, 1 -diyl)dicarbamate
Example 34A l-(4-fer?-butylphenyl)-2,5-bis(4-nitrophenyl)pyrrolidine The product from Example 1C (3.67 g, 7.51 mmol) and 4-tert-butylaniline (11.86 ml, 75 mmol) in DMF (40 ml) was stirred under nitrogen at 50 °C for 4 h. The resulting mixture was diluted into ethyl acetate, treated with IM HCl, stirred for 10 minutes and filtered to remove solids. The filtrate organic layer was washed twice with brine, dried with sodium sulfate, filtered and evaporated. The residue was purified by chromatography on silica gel eluting with ethyl acetate in hexane (5% to 30%) to give a solid. The solid was triturated in a minimal volume of 1 :9 ethyl acetate/hexane to give a light yellow solid as a mixture of trans and cis isomers (1.21 g, 36%).
Example 34B 4,4′-((2S,5S)-1-(4-tert-butylphenyl)pyrrolidine-2,5-diyl)dianiline and 4,4′-((2R,5R)-1-(4-fert- butylphenyl)pyrrolidine-2,5-diyl)dianiline To a solution of the product from Example 34A (1.1 g, 2.47 mmol) in ethanol (20 ml) and
THF (20 ml) was added PtC>2 (0.22 g, 0.97 mmol) in a 50 ml pressure bottle and stirred under 30 psi hydrogen at room temperature for 1 h. The mixture was filtered through a nylon membrane and evaporated. The residue was purified by chromatography on silica gel eluting with ethyl acetate in hexane (20% to 60%). The title compound eluted as the first of 2 stereoisomers (trans isomer, 0.51 g, 54%).
Example 34C
(2S,2’S)-tert-Butyl 2,2′-(4,4′-((2S,5S)-1-(4-fer/’-butylphenyl)pyrrolidine-2,5-diyl)bis(4,l- phenylene))bis(azanediyl)bis(oxomethylene)dipyrrolidine- 1 -carboxylate and (2S,2’S)-tert-Butyl 2,2′- (4,4′-((2R,5R)-1-(4-tert-butylphenyl)pyrrolidine-2,5-diyl)bis(4,l- phenylene))bis(azanediyl)bis(oxomethylene)dipyrrolidine-1-carboxylate To a mixture of the product from Example 34B (250 mg, 0.648 mmol), (S)-1-(tert- butoxycarbonyl)pyrrolidine-2-carboxylic acid (307 mg, 1.427 mmol) and HATU (542 mg, 1.427 mmol) in DMSO (10 ml) was added Hunig’s base (0.453 ml, 2.59 mmol). The reaction mixture was stirred at room temperature for 1 h. The mixture was partitioned with ethyl acetate and water. The organic layer was washed with brine, dried with sodium sulfate, filtered and evaporated. The residue was purified by chromatography on silica gel eluting with ethyl acetate in hexane (10% to 50%) to give the title compound (500 mg, 99%).
Example 34D
(2S,2’S)-N,N’-(4,4′-((2S,5S)-1-(4-ter/’-butylphenyl)pyrrolidine-2,5-diyl)bis(4,l- phenylene))dipyrrolidine-2-carboxamide and (2S,2’S)-N,N’-(4,4′-((2R,5R)-1-(4-tert- butylphenyl)pyrrolidine-2,5-diyl)bis(4,l-phenylene))dipyrrolidine-2-carboxamide To the product from Example 34C (498 mg, 0.638 mmol) in dichloromethane (4 ml) was added TFA (6 ml). The reaction mixture was stirred at room temperature for 1 h and concentrated in vacuo. The residue was partitioned between 3: 1 CHCl3dsopropyl alcohol and saturated aq. NaHCO3. The aqueous layer was extracted by 3: 1 CHCl3:isopropyl alcohol again. The combined organic layers were dried over
filtered and concentrated to give the title compound (345 mg, 93%).
Example 34E Dimethyl (2S,2’S)-l,r-((2S,2’S)-2,2′-(4,4′-((2S,5S)-1-(4-fert-butylphenyl)pyrrolidine-2,5-diyl)bis(4,l- phenylene))bis(azanediyl)bis(oxomethylene)bis(pyrrolidine-2, 1 -diyl))bis(3-methyl- 1 -oxobutane-2, 1 – diyl)dicarbamate and
Dimethyl (2S,2’S)-1, r-((2S,2’S)-2,2′-(4,4′-((2R,5R)-1-(4-fert-butylphenyl)pyrrolidine-2,5- diyl)bis(4, 1 -phenylene))bis(azanediyl)bis(oxomethylene)bis(pyrrolidine-2, 1 -diyl))bis(3-methyl- 1 – oxobutane-2, 1 -diyl)dicarbamate
The product from Example 34D (29.0 mg, 0.050 mmol), (S)-2-(methoxycarbonylamino)-3- methylbutanoic acid (19.27 mg, 0.110 mmol), EDAC (21.09 mg, 0.110 mmol), HOBT (16.85 mg,
0.110 mmol) and N-methylmorpholine (0.027 ml, 0.250 mmol) were combined in DMF (2 ml). The reaction mixture was stirred at room temperature for 3 h. The mixture was partitioned with ethyl acetate and water. The organic layer was washed with brine twice, dried with sodium sulfate, filtered and evaporated. The residue was purified by chromatography on silica gel eluting with ethyl acetate in hexane (50% to 80%) to give a solid. The solid was triturated with ethyl acetate/hexane to give the title compound (13 mg, 29%). 1H NMR (400 MHz, DMSO-D6) δ ppm 0.85 – 0.95 (m, 12 H) 1.11 (s, 9 H) 1.59 – 1.65 (m, 2 H) 1.79 – 2.04 (m, 8 H) 2.10 – 2.18 (m, 2 H) 2.41-2.46 (m, 2H) 3.52 (s, 6 H)
3.57 – 3.67 (m, 2 H) 3.76 – 3.86 (m, 2 H) 4.00 (t, J=7.56 Hz, 2 H) 4.39 – 4.46 (m, 2 H) 5.15 (d, J=7.00
Hz, 2 H) 6.17 (d, J=7.70 Hz, 2 H) 6.94 (d, J=8.78 Hz, 2 H) 7.13 (d, J=7.37 Hz, 4 H) 7.30 (d, J=8.20
Hz, 2 H) 7.50 (d, J=8.24 Hz, 4 H) 9.98 (s, 2 H); (ESI+) m/z 895 (M+H)+. The title compound showed an EC50 value of less than about 0.1 nM in HCV lb-Conl replicon assays in the presence of 5% FBS.
Example 35
Dimethyl (2S,2’S)-l,r-((2S,2’S)-2,2′-(4,4′-((2S,5S)-1-(4-tert-butylphenyl)pyrrolidine-2,5-diyl)bis(4,l- phenylene))bis(azanediyl)bis(oxomethylene)bis(pyrrolidine-2, 1 -diyl))bis(3-methyl- 1 -oxobutane-2, 1 – diyl)dicarbamate
The product from Example 34E was purified by chiral chromatography on a Chiralpak AD-H semi-prep column eluting with a 2:1 mixture of hexane:(2: l isopropyl alcohol: EtOH). The title compound was the first of the 2 diastereomers to elute. 1H NMR (400 MHz, DMSO-D6) δ ppm 0.88 (d, J=6.61 Hz, 6 H) 0.93 (d, J=6.72 Hz, 6 H) 1.11 (s, 9 H) 1.63 (d, J=5.42 Hz, 2 H) 1.80 – 2.04 (m, 8 H) 2.09 – 2.19 (m, 2 H) 2.44 – 2.47 (m, 2 H) 3.52 (s, 6 H) 3.59 – 3.66 (m, 2 H) 3.77 – 3.84 (m, 2 H) 4.02 (t, J=8.40 Hz, 2 H) 4.42 (dd, J=7.86, 4.83 Hz, 2 H) 5.14 (d, J=6.18 Hz, 2 H) 6.17 (d, J=8.67 Hz, 2 H) 6.94 (d, J=8.78 Hz, 2 H) 7.13 (d, J=8.46 Hz, 4 H) 7.31 (d, J=8.35 Hz, 2 H) 7.50 (d, J=8.35 Hz, 4 H) 9.98 (s, 2 H). The title compound showed an EC50 value of less than about 0.1 nM in HCV Ib- Conl replicon assays in the presence of 5% FBS.
Example 36 Dimethyl (2S,2’S)-1, r-((2S,2’S)-2,2′-(4,4′-((2R,5R)-1-(4-fert-butylphenyl)pyrrolidine-2,5- diyl)bis(4, 1 -phenylene))bis(azanediyl)bis(oxomethylene)bis(pyrrolidine-2, 1 -diyl))bis(3-methyl- 1 – oxobutane-2, 1 -diyl)dicarbamate
The product from Example 34E was purified by chiral chromatography on a Chiralpak AD-H semi-prep column eluting with a 2:1 mixture of hexane:(2: l isopropyl alcohol: EtOH). The title compound was the second of 2 diastereomers to elute. 1H NMR (400 MHz, DMSO-D6) δ ppm 0.87
(d, J=6.51 Hz, 6 H) 0.92 (d, J=6.72 Hz, 6 H) 1.11 (s, 9 H) 1.63 (d, J=5.53 Hz, 2 H) 1.82 – 2.04 (m, 8
H) 2.09-2.18 (m, 2 H) 2.41 – 2.47 (m, 2 H) 3.52 (s, 6 H) 3.58 – 3.67 (m, 2 H) 3.75 – 3.84 (m, 2 H) 4.02
(t, J=7.26 Hz, 2 H) 4.43 (dd, J=7.92, 4.88 Hz, 2 H) 5.14 (d, J=6.18 Hz, 2 H) 6.17 (d, J=8.78 Hz, 2 H) 6.94 (d, J=8.67 Hz, 2 H) 7.12 (d, J=8.46 Hz, 4 H) 7.31 (d, J=8.35 Hz, 2 H) 7.49 (d, J=8.46 Hz, 4 H)
9.98 (s, 2 H). The title compound showed an EC50 value of less than about 0.1 nM in HCV lb-Conl replicon assays in the presence of 5% FBS.
Example 37 Dimethyl (2S,2’S)-l,r-((2S,2’S)-2,2′-(4,4′-((2S,5S)-1-(4-fert-butylphenyl)pyrrolidine-2,5-diyl)bis(4,l- phenylene))bis(azanediyl)bis(oxomethylene)bis(pyrrolidine-2, 1 -diyl))bis(3-methyl- 1 -oxobutane-2, 1 – diyl)dicarbamate
Example 37A (S)-2,5-dioxopyrrolidin-1-yl 2-(methoxycarbonylamino)-3-methylbutanoate To a mixture of (S)-2-(methoxycarbonylamino)-3-methylbutanoic acid (19.66 g, 112 mmol) and N-hydroxysuccinimide (13.29g, 116 mmol) was added ethyl acetate (250 ml), and the mixture was cooled to 0-5 °C. Diisopropylcarbodiimide (13.88 g, 110 mmol) was added and the reaction mixture was stirred at 0-5 °C for about 1 hour. The reaction mixture was warmed to room temperature. The solids (diisopropylurea by-product) were filtered and rinsed with ethyl acetate. The filtrate was concentrated in vacuo to an oil. Isopropyl alcohol (200 ml) was added to the oil and the mixture was heated to about 50 °C to obtain a homogeneous solution. Upon cooling, crystalline solids formed. The solids were filtered and washed with isopropyl alcohol (3 x 20 ml) and dried to give the title compound as a white solid (23.2 g, 77% yield).
Example 37B
(S)- 1 -((S)-2-(methoxycarbonylamino)-3-methylbutanoyl)pyrrolidine-2-carboxylic acid To a mixture of L-proline (4.44g, 38.6 mmol), water (20 ml), acetonitrile (20 ml) and DIEA (9.5 g, 73.5 mmol) was added a solution of the product from Example 37A (1Og, 36.7 mmol) in acetonitrile (20 inL) over 10 minutes. The reaction mixture was stirred overnight at room temperature. The solution was concentrated under vacuum to remove the acetonitrile. To the resulting clear water solution was added 6N HCl (9 ml) until pH ~ 2 .The solution was transferred to a separatory funnel and 25% NaCl (10 ml) was added and the mixture was extracted with ethyl acetate (75 ml), and then again with ethyl acetate (6 x 20 ml), and the combined extracts were washed with 25% NaCl (2 x 10ml). The solvent was evaporated to give a thick oil. Heptane was added and the solvent was evaporated to give a foam, which was dried under high vacuum. Diethyl ether was added and the solvent was evaporated to give a foam, which was dried under high vacuum to give the title compound (10.67g) as a white solid.
The compound of Example 37B can also be prepreared according to the following procedure: To a flask was charged L- valine (35 g, 299 mmol), IN sodium hydroxide solution (526 ml,
526 mmol) and sodium carbonate (17.42 g, 164 mmol). The mixture was stirred for 15 min to dissolve solids and then cooled to 15 °C. Methyl chloroformate (29.6 g, 314 mmol) was added slowly to the reaction mixture. The mixture was then stirred at rt for 30 min. The mixture was cooled to 15 °C and pH adjusted to -5.0 with concentrated HCl solution. 100 inL of 2-methytetrahydrofuran (2- MeTHF) was added and the adjustment of pH continued until the pH reached ~ 2.0. 150 mL of 2- MeTHF was added and the mixture was stirred for 15 min. Layers were separated and the aqueous layer extracted with 100 mL of 2-MeTHF. The combined organic layer was dried over anhyd Na2SC^ and filtered, and Na2SC^ cake was washed with 50 mL of 2-MeTHF. The product solution was concentrated to ~ 100 mL, chased with 120 mL of IPAc twice. 250 mL of heptanes was charged slowly and then the volume of the mixture was concentrated to 300 mL. The mixture was heated to 45 °C and 160 mL of heptanes charged. The mixture was cooled to rt in 2h, stirred for 30 min, filtered and washed with 2-MeTHF/heptanes mixture (1:7, 80 inL). The wetcake was dried at 55 °C for 24 h to give 47.1 g of Moc-L- VaI-OH product as a white solid (90%).
Moc-L- VaI-OH (15O g, 856 mmol), HOBt hydrate (138 g, 899 mmol) and DMF (1500 ml) were charged to a flask. The mixture was stirred for 15 min to give a clear solution. EDC hydrochloride (172 g, 899 mmol) was charged and mixed for 20 min. The mixture was cooled to 13
°C and (L)-proline benzyl ester hydrochloride (207 g, 856 mmol) charged. Triethylamine (109 g,
1079 mmol) was then charged in 30 min. The resulting suspension was mixed at rt for 1.5 h. The reaction mixture was cooled to 15 °C and 1500 mL of 6.7% NaHCO3 charged in 1.5 h, followed by the addition of 1200 mL of water over 60 min. The mixture was stirred at rt for 30 min, filtered and washed with water/DMF mixture (1 :2, 250 mL) and then with water (1500 mL). The wetcake was dried at 55 °C for 24 h to give 282 g of product as a white solid (90%).
The resulting solids (40 g) and 5% Pd/ Alumina were charged to a Parr reactor followed by THF (160 mL). The reactor was sealed and purged with nitrogen (6 x 20 psig) followed by a hydrogen purge (6 x 30 psig). The reactor was pressurized to 30 psig with hydrogen and agitated at room temperature for approximately 15 hours. The resulting slurry was filtered through a GF/F filter and concentrated to approximately 135 g solution. Heptane was added (120 mL), and the solution was stirred until solids formed. After an addition 2 – 3 hours additional heptane was added drop-wise (240 mL), the slurry was stirred for approximately 1 hour, then filtered. The solids were dried to afford the title compound.
Example 37C
(lR,4R)-1,4-bis(4-nitrophenyl)butane-1,4-diyl dimethanesulfonate
The product from Example 32 (5.01 g, 13.39 mmol) was combined with 2- methyltetrahydrofuran (70 mL) and cooled to -5 °C, and N,N-diisopropylethylamine (6.81 g, 52.7 mmol) was added over 30 seconds. Separately, a solution of methanesulfonic anhydride (6.01 g, 34.5 mmol) in 2-methyltetrahydrofuran (30 mL) was prepared and added to the diol slurry over 3 min., maintaining the internal temperature between -15 °C and -25 °C. After mixing for 5 min at -15 °C, the cooling bath was removed and the reaction was allowed to warm slowly to 23 °C and mixed for 30 minutes. After reaction completion, the crude slurry was carried immediately into the next step.
Example 37D
(2S,5S)-1-(4-tert-butylphenyl)-2,5-bis(4-nitrophenyl)pyrrolidine
To the crude product solution from Example 37C (7.35 g, 13.39 mmol) was added 4-tert- butylaniline (13.4 g, 90 mmol) at 23 °C over 1 minute. The reaction was heated to 65 °C for 2 h. After completion, the reaction mixture was cooled to 23 °C and diluted with 2-methyltetrahydrofuran (100 mL) and 1 M HCl (150 mL). After partitioning the phases, the organic phase was treated with 1 M HCl (140 mL), 2-methyltetrahydrofuran (50 mL), and 25 wt% aq. NaCl (100 mL), and the phases were partitioned. The organic phase was washed with 25 wt% aq. NaCl (50 mL), dried over MgSO/t, filtered, and concentrated in vacuo to approximately 20 mL. Heptane (30 mL) and additional 2- methyltetrahydrofuran were added in order to induce crystallization. The slurry was concentrated further, and additional heptane (40 mL) was slowly added and the slurry was filtered, washing with 2- methyltetrahydrofuran:heptane (1:4, 20 mL). The solids were suspended in MeOH (46 mL) for 3 h, filtered, and the wet solid was washed with additional MeOH (18 mL). The solid was dried at 45 °C in a vacuum oven for 16 h to provide the title compound (3.08 g, 51% 2-step yield).
Example 37E
4,4′-((2S,5S)-1-(4-tert-butylphenyl)pyrrolidine-2,5-diyl)dianiline
To a 160 ml Parr stirrer hydrogenation vessel was added the product from Example 37D (2 g, 4.49 mmol), followed by 60 ml of THF, and Raney Nickel Grace 2800 (1 g, 50 wt% (dry basis)) under a stream of nitrogen. The reactor was assembled and purged with nitrogen (8 x 20 psig) followed by purging with hydrogen (8 x 30 psig). The reactor was then pressurized to 30 psig with hydrogen and agitation (700 rpm) began and continued for a total of 16 h at room temperature. The slurry was filtered by vacuum filtration using a GF/F Whatman glass fiber filter. Evaporation of the filtrate to afford a slurry followed by the addition heptane and filtration gave the crude title compound, which was dried and used directly in the next step.
Example 37F dimethyl (2S,2’S)-l,r-((2S,2’S)-2,2′-(4,4′-((2S,5S)-1-(4-tert-butylphenyl)pyrrolidine-2,5-diyl)bis(4, l- phenylene)bis(azanediyl)bis(oxomethylene))bis(pyrrolidine-2, 1 -diyl))bis(3-methyl- 1 -oxobutane-2, 1 – diy 1) die arb amate To a solution of the product from Example 37E (1.64 g, 4.25 mmol) in DMF (20 ml), the product from Example 37B (2.89 g, 10.63 mmol), and HATU (4.04 g, 10.63 mmol) in DMF (15OmL) was added triethylamine (1.07 g, 10.63 mmol), and the solution was stirred at room temperature for 90 min. To the reaction mixture was poured 20 mL of water, and the white precipitate obtained was filtered, and the solid was washed with water (3×5 mL). The solid was blow dried for Ih. The crude material was loaded on a silica gel column and eluted with a gradient starting with ethyl acetate/ heptane (3/7), and ending with pure ethyl acetate. The desired fractions were combined and solvent distilled off to give a very light yellow solid, which was dried at 45 °C in a vacuum oven with nitrogen purge for 15 h to give the title compound (2.3 g, 61% yield). 1H NMR (400 MHz, DMSO- D6) δ ppm 0.88 (d, J=6.61 Hz, 6 H) 0.93 (d, J=6.72 Hz, 6 H) 1.11 (s, 9 H) 1.63 (d, J=5.42 Hz, 2 H) 1.80 – 2.04 (m, 8 H) 2.09 – 2.19 (m, 2 H) 2.44 – 2.47 (m, 2 H) 3.52 (s, 6 H) 3.59 – 3.66 (m, 2 H) 3.77 – 3.84 (m, 2 H) 4.02 (t, J=8.40 Hz, 2 H) 4.42 (dd, J=7.86, 4.83 Hz, 2 H) 5.14 (d, J=6.18 Hz, 2 H) 6.17 (d, J=8.67 Hz, 2 H) 6.94 (d, J=8.78 Hz, 2 H) 7.13 (d, J=8.46 Hz, 4 H) 7.31 (d, J=8.35 Hz, 2 H) 7.50 (d, J=8.35 Hz, 4 H) 9.98 (s, 2 H).
Alternately, the product from example 37E (11.7 g, 85 wt%, 25.8 mmol) and the product from example 37B (15.45 g, 56.7 mmol) are suspended in EtOAc (117 mL), diisopropylethylamine (18.67 g, 144 mmol) is added and the solution is cooled to 0 °C. In a separate flask, 1-propanephosphonic acid cyclic anhydride (T3P®) (46.0 g, 50 wt% in EtOAc, 72.2 mmol) was dissolved in EtOAc (58.5 mL), and charged to an addition funnel. The T3P solution is added to the reaction mixture drop-wise over 3-4 h and stirred until the reaction is complete. The reaction is warmed to room temperature,and washed with IM HCl/7.5 wt% NaCl (100 mL), then washed with 5% NaHCO3 (100 mL), then washed with 5% NaCl solution (100 mL). The solution was concentrated to approximately 60 mL, EtOH (300 mL) was added, and the solution was concentrated to 84 g solution.
A portion of the EtOH solution of product (29 g) was heated to 40 °C, and added 134 g 40 w% EtOH in H2O. A slurry of seeds in 58 wt/wt% EtOH/H2O was added, allowed to stir at 40 °C for several hours, then cooled to 0 °C. The slurry is then filtered, and washed with 58wt/wt% EtOH/H2O. The product is dried at 40 – 60 °C under vacuum, and then rehydrated by placing a tray of water in the vacuum oven to give the title compound. The title compound showed an EC50 value of less than about 0.1 nM in HCV lb-Conl replicon assays in the presence of 5% FBS.
Intermediates
Example 32
( 1 R,4R)- 1 ,4-bis(4-mtrophenyl)butane- 1 ,4-diol
To (S)-(-)-α,α-diphenyl-2-pyrrohdinemethanol (2 71 g, 10 70 mmol) was added THF (80 mL) at 23 °C The very thin suspension was treated with t11methyl borate (1 44 g, 13 86 mmol) over 30 seconds, and the resulting solution was mixed at 23 °C for 1 h The solution was cooled to 16-19 °C, and N,N-diethylanilme borane (21 45 g, 132 mmol) was added dropwise via syringe over 3-5 mm (caution vigorous H2 evolution), while the internal temperature was maintained at 16-19 °C After 15 mm, the H2 evolution had ceased To a separate vessel was added the product from Example IA (22 04 g, 95 wt%, 63 8 mmol), followed by THF (80 mL), to form an orange slurry After cooling the slurry to 11 °C, the borane solution was transferred via cannula into the dione slurry over 3-5 min During this period, the internal temperature of the slurry rose to 16 °C After the addition was complete, the reaction was maintained at 20-27 °C for an additional 2 5 h After reaction completion, the mixture was cooled to 5 °C and methanol (16 7 g, 521 mmol) was added dropwise over 5-10 mm, maintaining an internal temperature <20 °C (note vigorous H2 evolution) After the exotherm had ceased (ca 10 mm), the temperature was adjusted to 23 °C, and the reaction was mixed until complete dissolution of the solids had occurred Ethyl acetate (300 mL) and 1 M HCl (120 mL) were added, and the phases were partitioned The organic phase was then washed successively with 1 M HCl (2 x 120 mL), H2O (65 mL), and 10% aq NaCl (65 mL) The orgamcs were dried over MgSO4, filtered, and concentrated in vacuo Crystallization of the product occurred during the concentration The slurry was warmed to 50 °C, and heptane (250 inL) was added over 15 min. The slurry was then allowed to mix at 23 °C for 30 min and filtered. The wet cake was washed with 3: 1 heptane:ethyl acetate (75 mL), and the orange, crystalline solids were dried at 45 °C for 24 h to provide the title compound (15.35 g, 99.3% ee, 61% yield), which was contaminated with 11% of the meso isomer (vs. dl isomer).
 |
|
| Systematic (IUPAC) name | |
|---|---|
|
Dimethyl ({(2S,5S)-1-[4-(2-methyl-2-propanyl)phenyl]-2,5-pyrrolidinediyl}bis{4,1-phenylenecarbamoyl(2S)-2,1-pyrrolidinediyl[(2S)-3-methyl-1-oxo-1,2-butanediyl]})biscarbamate
|
|
| Clinical data | |
| Trade names | Viekira Pak (with ombitasvir, paritaprevir, ritonavir and dasabuvir), Technivie (with ombitasvir, paritaprevir, and ritonavir) |
| Legal status |
|
| Routes of administration |
Oral |
| Pharmacokinetic data | |
| Bioavailability | not determined |
| Protein binding | ~99.9% |
| Metabolism | amide hydrolysis followed by oxidation |
| Onset of action | ~4 to 5 hours |
| Biological half-life | 21 to 25 hours |
| Excretion | mostly with feces (90.2%) |
| Identifiers | |
| CAS Registry Number | 1258226-87-7 |
| PubChem | CID: 54767916 |
| ChemSpider | 31136214 |
| ChEBI | CHEBI:85183 |
| Synonyms | ABT-267 |
| Chemical data | |
| Formula | C50H67N7O8 |
| Molecular mass | 894.11 g/mol |
rx list
VIEKIRA PAK is ombitasvir, paritaprevir, ritonavir fixed dose combination tablets copackaged with dasabuvir tablets.
Ombitasvir, paritaprevir, ritonavir fixed dose combination tablet includes ahepatitis C virus NS5A inhibitor (ombitasvir), a hepatitis C virus NS3/4Aprotease inhibitor (paritaprevir), and a CYP3A inhibitor (ritonavir) that inhibits CYP3A mediated metabolism of paritaprevir, thereby providing increased plasma concentration of paritaprevir. Dasabuvir is a hepatitis C virus nonnucleoside NS5B palm polymerase inhibitor, which is supplied as separate tablets in the copackage. Both tablets are for oral administration.
The chemical name of ombitasvir is Dimethyl ([(2S,5S)-1-(4-tert-butylphenyl) pyrrolidine-2,5diyl]bis{benzene-4,1-diylcarbamoyl(2S)pyrrolidine-2,1-diyl[(2S)-3-methyl-1-oxobutane-1,2diyl]})biscarbamate hydrate. The molecular formula is C50H67N7O8•4.5H2O (hydrate) and the molecular weight for the drug substance is 975.20 (hydrate). The drug substance is white to light yellow to light pink powder, and is practically insoluble in aqueous buffers but is soluble in ethanol. Ombitasvir has the following molecular structure:


The chemical name of paritaprevir is (2R,6S,12Z,13aS,14aR,16aS)-N-(cyclopropylsulfonyl)-6{[(5-methylpyrazin-2-yl)carbonyl]amino}-5,16-dioxo-2-(phenanthridin-6-yloxy)1,2,3,6,7,8,9,10,11,13a,14,15,16,16a-tetradecahydrocyclopropa[e]pyrrolo[1,2-a][1,4] diazacyclopentadecine-14a(5H)-carboxamide dihydrate. The molecular formula is C40H43N7O7S•2H2O (dihydrate) and the molecular weight for the drug substance is 801.91 (dihydrate). The drug substance is white to off-white powder with very low water solubility. Paritaprevir has the following molecular structure:
 |
The chemical name of ritonavir is [5S-(5R*,8R*,10R*,11R*)]10-Hydroxy-2-methyl-5-(1methyethyl)-1-[2-(1-methylethyl)-4-thiazolyl]-3,6-dioxo-8,11-bis(phenylmethyl)-2,4,7,12tetraazatridecan-13-oic acid,5-thiazolylmethyl ester. The molecular formula is C37H48N6O5S2 and the molecular weight for the drug substance is 720.95. The drug substance is white to off white to light tan powder practically insoluble in water and freely soluble in methanol and ethanol. Ritonavir has the following molecular structure:
Ombitasvir, paritaprevir, and ritonavir film-coated tablets are co-formulated immediate release tablets. The tablet contains copovidone, K value 28,vitamin E polyethylene glycol succinate, propylene glycol monolaurate Type I, sorbitan monolaurate, colloidal silicon dioxide/colloidal anhydrous silica, sodium stearyl fumarate, polyvinyl alcohol, polyethylene glycol 3350/macrogol 3350, talc, titanium dioxide, and iron oxide red. The strength for the tablet is 12.5 mg ombitasvir, 75 mg paritaprevir, 50 mg ritonavir.
The chemical name of dasabuvir is Sodium 3-(3-tert-butyl-4-methoxy-5-{6[(methylsulfonyl)amino]naphthalene-2-yl}phenyl)-2,6-dioxo-3,6-dihydro-2H-pyrimidin-1-ide hydrate (1:1:1). The molecular formula is C26H26N3O5S•Na•H2O (salt, hydrate) and the molecular weight of the drug substance is 533.57 (salt, hydrate). The drug substance is white to pale yellow to pink powder, slightly soluble in water and very slightly soluble in methanol and isopropyl alcohol. Dasabuvir has the following molecular structure:
 |
Dasabuvir is formulated as a 250 mg film-coated, immediate release tablet containing microcrystalline cellulose (D50-100 um), microcrystalline cellulose (D50-50 um), lactose monohydrate, copovidone, croscarmellose sodium, colloidal silicon dioxide/anhydrous colloidal silica, magnesium stearate, polyvinyl alcohol, titanium dioxide, polyethylene glycol 3350/macrogol 3350, talc, and iron oxide yellow, iron oxide red and iron oxide black. Each tablet contains 270.3 mg dasabuvir sodium monohydrate equivalent to 250 mg dasabuvir.
//////////fda 2014, Ombitasvir, orphan drug, Abbvie Inc.












































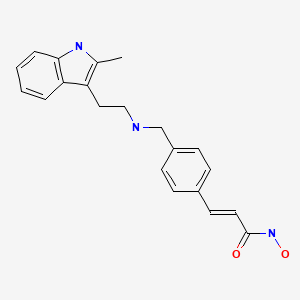 panobinostat
panobinostat


















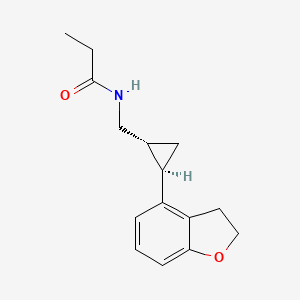




































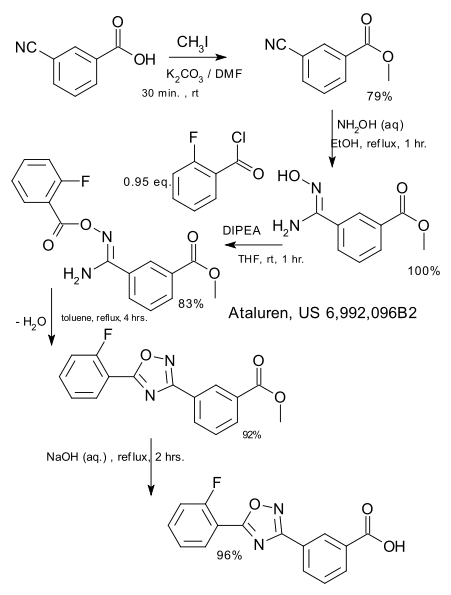





























































































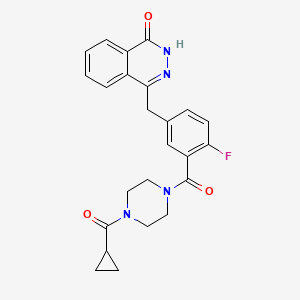
































 Bafetinib
Bafetinib